
X-viazaná adrenoleukodystrofia
X-adrenoleukodystrophy
Introduction:
X-adrenoleukodystrophy (X-ALD) is the most common peroxisomal disorder characterized by adrenal insufficiency and neurological manifestations. The disorder is caused by a defect in the ABCD1 gene, leading to inability to degrade very long chain fatty acids (VLCFA) and to their accumulation in tissues and fluids. The disorder is recessively inherited and X-linked with clinical manifestation predominantly in males. Clinical manifestation includes several phenotypes of X-ALD. The aim of the study was to review current knowledge on X-ALD and our options for laboratory, radiological and genetic diagnosis of the disorder and treatment approaches.
Material and methods:
Our study group included 11 patients, five with childhood cerebral form, one with the adolescent cerebral form, two with primary adrenal insufficiency, one with andrenomyeloneuropathy and two symptomatic females. All patients underwent VLCFA testing, brain MRI and DNA analysis of the ABCD1 gene.
Results:
Pathologically elevated VLCFA plasma levels were found in all patients. MRI revealed changes in white matter typically distributed in parietal-occipital areas in all but one patient. Mutation of the ABCD1 gene was confirmed in all patients. All male patients were treated with Lorenzo´s oil. Two patients with cerebral form died within 2 years since the clinical onset of the disease. One patient was successfully transplanted (sibling transplantation of umbilical cord blood).
Conclusion:
The birth rate in Slovakia is about 60 thousand children per year, and the incidence of the X-ALD is from 1 : 16,800 to 1 : 42,000. Therefore, we presume that there will be one to three new cases each year. It is important to recognize the disorder from its typical clinical manifestationa. Due to the close relationship between the disorder and endocrinopathies, education of endocrinologists is considered important, in order to increase their ability to identify possible cases without neurological deficit. Cerebral forms of the disease in their early stages can be succesfully treated with bone marrow or umbilical cord blood transplantation.
Key words:
X-adrenoleukodystrophy – classification – pathogenesis – diagnosis – treatment
Authors:
M. Kolníková 1; P. Sýkora 1; R. Petrovič 2; M. Fischerová 2; J. Chandoga 2
Authors‘ workplace:
Klinika detskej neurológie LF UK a DFNsP Bratislava
1; Ústav lekárskej biológie, genetiky a klinickej genetiky LF UK a UN Bratislava, oddelenie molekulárnej a biochemickej genetiky, FNsP Bratislava-Staré Mesto
2
Published in:
Cesk Slov Neurol N 2013; 76/109(2): 197-202
Category:
Short Communication
Overview
Úvod:
X-viazaná adrenoleukodystrofia (X-ALD) je najčastejšia peroxizómová porucha charakterizovaná adrenálnou insuficienciou a neurologickou manifestáciou. Typ dedičnosti je gonozómovo recesívny, preto sa manifestuje hlavne u mužského pohlavia. Je spôsobená defektom v ABCD1 géne. Zmena génu vedie k poruche odbúravania veľmi dlhých mastných kyselín (VLCFA) a následne k ich hromadeniu v tkanivách i tekutinách. Podľa klinického obrazu je niekoľko fenotypov X-ALD. Cieľ práce bol priblížiť súčasný stav poznatkov o X-ALD, naše možnosti laboratórnej, rádiologickej a genetickej diagnostiky ochorenia a liečebné postupy.
Pacienti a metóda:
Do súboru sme začlenili 11 pacientov, päť s detskou cerebrálnou formou, jedného s cerebrálnou adolescentnou formou, dvoch s primárnou adrenálnou insuficienciou, jedného s adrenomyeloneuropatiou a dve symptomatické ženy. Pacienti mali vyšetrené VLCFA v plazme, MR mozgu a DNA analýzu ABCD1 génu.
Výsledky:
U všetkých pacientov bol nález patologicky zvýšených hodnôt VLCFA v plazme. Na MR mozgu sme okrem jedného pacienta našli typické zmeny v bielej hmote parieto-okcipitálne. Všetci pacienti mali nález mutácie v ABCD1 géne. Dvaja pacienti s cerebrálnou formou zomreli do dvoch rokov od začiatku ochorenia. Piati pacienti mali liečbu Lorenzovým olejom. Jeden pacient bol úspešne transplantovaný súrodeneckou transplantáciou a jeden pacient je aktuálne po transplantácii.
Záver:
Pri pôrodnosti na Slovensku 60 000 detí ročne a incidencii ochorenia 1 : 16 800 až 1 : 42 000 predpokladáme, že môže pribudnúť jeden až tri nové prípady ročne. Dôležité je rozpoznať typický klinický obraz ochorenia. Pre úzky vzťah X-ALD a endokrinopatie je významná tiež edukácia endokrinológov, kedy je možné zachytiť prípady bez neurologickej symptomatológie. Detskú a adolescentnú cerebrálnu formu je možné liečiť vo včasnom štádiu transplantáciou kostnej drene alebo pupočníkovej krvi.
Kľúčové slová:
X-adrenoleukodystrofia – klasifikácia – patogenéza – diagnostika – liečba
Úvod
Biochemická podstata a formy
Muži s X-ALD majú od narodenia vyššie hodnoty VLCFA, čo je charakteristický znak ochorenia. VLCFA sú nasýtené mastné kyseliny s reťazcom dlhším ako 22 uhlíkov. Tento nález nie je špecifický pre X-ALD, je obrazom aj iných ochorení s poruchou peroxizómovej beta-oxidácie. Nedostatočne odbúravané VLCFA spôsobujú v CNS a PNS vznik veľmi nestabilného myelínu [1,2].
Aj keď akumulácia VLCFA je vo všetkých tkanivách organizmu, postihnutý je len kortex nadobličky a nervový systém. Zmeny, ktoré vznikajú, sú veľmi variabilné čo do začiatku veku, ale i manifestácie (tab. 1).
![Klinická variabilita X-ALD u mužov (podľa [1]).](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image/eb572e267bccfe416c21bceaba60342d.png)
Detská cerebrálna adrenoleukodystrofia je najznámejšia zo všetkých a dlho bola jedinou rozpoznávanou formou. Postihuje chlapcov s normálnym vývojom v predchorobí. Vekový priemer je zvyčajne medzi 4. a 10. rokom s najvyšším výskytom okolo 7. ± 1,7. roku života. Začiatok je nenápadný – prítomná je porucha pozornosti a diskrétne zmeny v správaní, ktoré sa môžu mylne hodnotiť ako ADHD. Postupne sa objavujú neurologické príznaky a epileptické záchvaty, ktoré môžu byť prvým príznakom.
Významný je obraz MR zmien bielej hmoty mozgu. Prítomné sú bilaterálne symetrické hyperintenzívne lézie na T2V a FLAIR obraze a súčasne hypointezívne zmeny na T1V obraze. Tento nález je v 85 % parieto-okcipitálne tzv. zadná leukodystrofia.
Ochorenie postupuje rýchlo, vzniká kortikálna slepota, strata sluchu, spastická kvadruparéza so stratou motorických schopností a prehĺtania. Čas od prvých príznakov do plne rozvinutej choroby je krátky, 6–12 mesiacov. Chlapci zomierajú zvyčajne do dvoch rokoch od začiatku ochorenia.
Adrenomyeloneuropatia (AMN) je definovaná ako ochorenie dospelých, neurologické postihnutie je spôsobené primárne myelopatiou a menej výrazne neuropatiou. Degeneráciou sú postihnuté najviac kortikospinálne dráhy a zadné povrazce, môže byť tiež obraz distálnej polyneuropatie s poruchou vibračnej citlivosti na dolných končatinách. AMN je najčastejšou neurologickou manifestáciou, postihujúcou približne 65 % rizikových mužov, a je často nediagnostikovaná. Pre občasné nálezy demyelinizačných lézií na MR mozgu je zamieňaná za SM. Začína medzi 14. a 60. rokom veku ako pomaly postupujúca progresívna spastická paraparéza, distálne senzorické poruchy, inkontinencia a sexuálna dysfunkcia. Zvyčajne je bez postihnutia mozgu. Asi dve tretiny pacientov má adrenálnu insuficienciu. Na EMG môžu byť spomalené rýchlosti vedenia. MR zobrazuje atrofiu miechy. Zmeny na MR mozgu sa popisujú podľa niektorých prác od 19 do 40 % prípadov a môžu ďalej progredovať tak ako zmeny u cerebrálnej formy.
Adolescentná a adultná cerebrálna forma sú podobné detskej cerebrálnej forme, ale priebeh je pomalší. Zmeny na mieche sú minimálne. Spinocerebelárna forma sa radí ako variant cerebrálnej AMN.
Primárna adrenálna insuficiencia alebo Addisonova choroba môže vznikať (ako forma X-ALD) v každom veku a nesúvisí s objavením sa neurologických príznakov. Dochádza k deficitu glukokortikoidov a minerálokortikoidov so zvýšením hodnôt ACTH a znížením kortizolu. Forma primárnej adrenálnej insuficiencie je odhadovaná na 35 % pacientov s X-ALD a až 70 % X-ALD má v priebehu ochorenia rozvoj nadobličkovej nedostatočnosti. Ochorenie sa môže manifestovať hypotenziou a hypoglykémiou pri ľahkých infekciách, pričom nerozpoznaná adrenálna kríza sa môže končiť fatálne. Vek vzniku je variabilný. Najmladší uvedený kojenec, kde sa popisuje adrenálne zlyhanie, bol vo veku šiestich mesiacov.
Asymptomatické prípady sú známe ako prípady so zvýšenými hodnotami VLCFA bez neurologickej a nadobličkovej patológie v priebehu celého života.
Heterozygotná forma u žien, ktoré sú prenášačky ochorenia v detskom veku a nemajú príznaky, avšak môžu mať myelopatické a neuropatické zmeny v dospelom veku. Viac ako 50 % týchto žien má ťažkosti s chôdzou, pridružujú sa dysestézie a problémy s močením. Zriedkavý je vývoj adrenálnej insuficiencie a cerebrálneho postihnutia, takže sa vyšetrenie MR a adrenálnych funkcií nevyžaduje [1–4].
Diagnóza
Hlavnou diagnostickou metódou je vyšetrenie hladiny VLCFA v plazme, fibroblastoch alebo tkanive. Rutinne sa používa vyšetrenie plazmy. Stanovuje sa hodnota C26:0 (kyselina cerotová), ako i pomer C26:0/C22:0 (kyelina cerotová : : kyselina behénová) a C24:0/C22:0 (kyselina lignicerová : kyselina behénová). Väčšina pacientov má zvýšené všetky tri zložky. Len u malého počtu nájdeme abnormnú hodnotu jedného alebo dvoch parametrov. U dojčiat s X-ALD sa zvyšuje hladina VLCFA v prvé dni života, dokonca sa môže nájsť zvýšenie už v pupočníkovej krvi. Včasné symptómy cerebrálnej formy X-ALD chlapcov je ťažké odlíšiť od poruchy pozornosti charakteru ADHD a ani nie je možné robiť skríning všetkým chlapcom s poruchou pozornosti. Vyšetrenie sa robí vtedy, keď sa objavia nezvyčajné príznaky a progresia stavu. Zmeny na MR mozgu sa začínajú v spleniu corpus callosum a postupne sa rozširujú do parieto--okcipitálnych oblastí bielej hmoty. Typický je obraz parieto-okcipitálnych zmien bielej hmoty charakteru zadnej leukodystrofie (obr. 1, 2). Ochorenie môže začínať aj zmenami frontálne, jednostranne asymetricky (skôr u dospelých) a môžu byť postihnuté zraková a sluchová dráha, pyramidová dráha, bazálne gangliá, thalamus a cerebelum. Podaním kontrastu na MR je možný dôkaz aktivity lézie (girlandovitý okraj lézie).
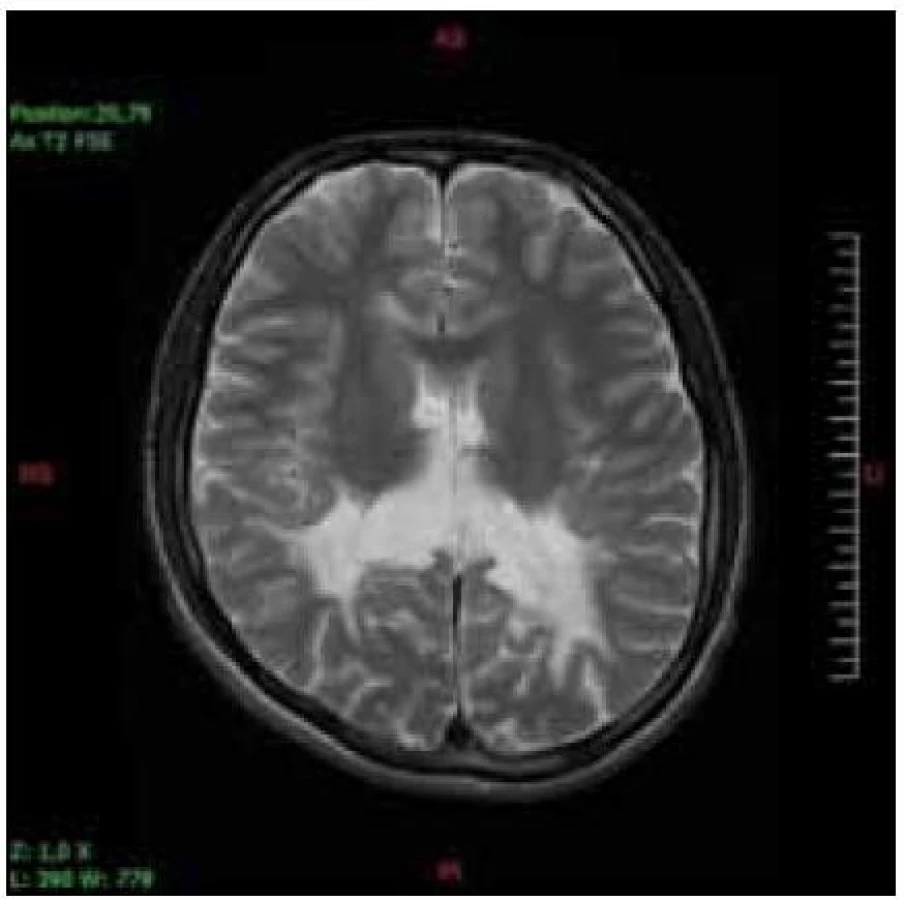
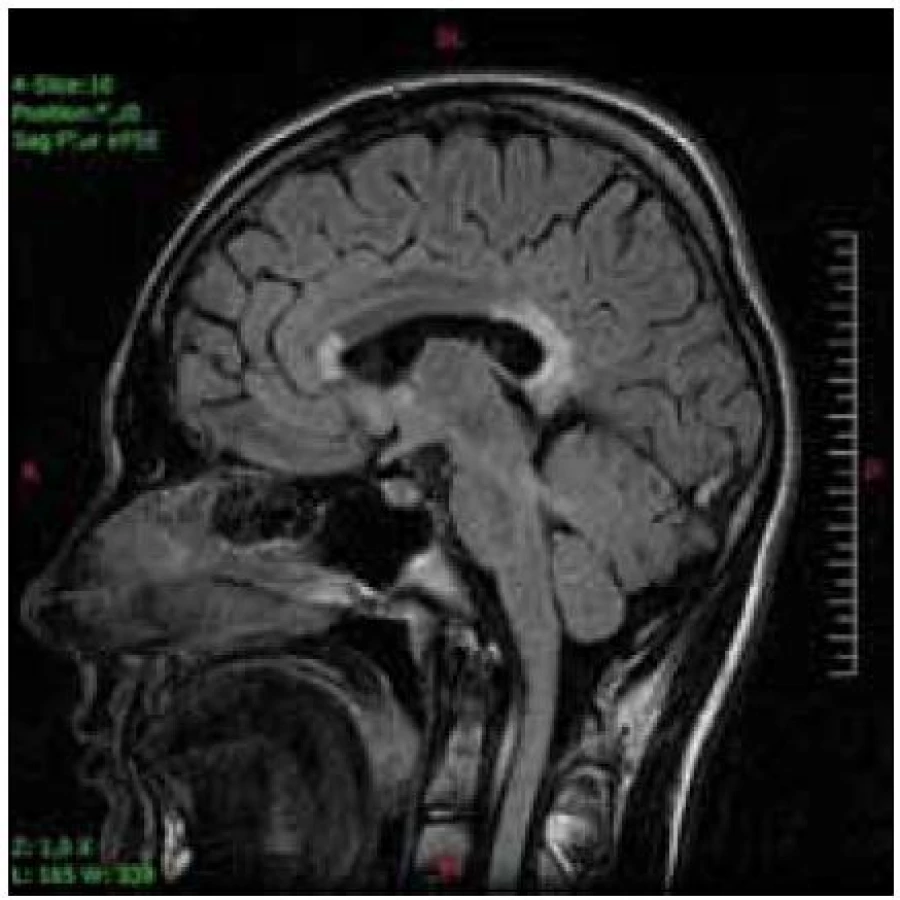
Dôležité je laboratórne vyšetrenie ostatných členov rodiny vrátane prenášačiek, čím môžu byť zachytený pacienti vo včasnom štádiu a asymptomatickí pacienti. Pre nosičky je v prípade negatívnych hodnôt VLCFA doporučená DNA analýza na potvrdenie diagnózy.
Prenatálna diagnostika plodu sa robila v minulosti vyšetrením VLCFA v choriových klkoch. Dnes sa zistí mutácia pomocou sekvenčnej analýzy u postihnutých príslušníkov a u matiek sa robí selektívne molekulové genetické vyšetrenie amniocytov. Perspektívne je použitie skríningu novorodencov zo suchej kvapky, tandemovou hmotnostnou spektrometriou na frakciu fosfatidil cholínu [1,5,6].
Patogenéza
Poznanie patogenézy X-ALD a jej rôznorodej neurologickej symptomatológie nie je kompletné. Princípom dvoch rôznych klinických obrazov ochorenia je nezápalové axonálne poškodenie periférnych nervov a miechy, verzus zápalové postihnutie bielej hmoty mozgu. Je pravdepodobné, že axonálne postihnutie, aké vidíme u AMN, je vlastne primárne poškodenie pri adrenoleukodystrofii, avšak 50 % chlapcov zomiera na zápalové postihnutie mozgu ešte pred manifestáciou axonopatie. Túto hypotézu podporujú modelové štúdie na myšiach [7–9] aj post mortem nálezy u ľudí [1]. Podľa súčasných názorov je podstatou procesov excesívna akumulácia VLCFA v tkanivách. VLCFA s väčšou dĺžkou alifatických reťazcov spôsobujú insolubilitu, čím sa narušuje membránová štruktúra a poškodzujú sa fyziologické vlastnosti rozličných bunkových funkcií. Vysoký výskyt VLCFA v myelíne poškodzuje stabilitu myelínového puzdra. Predpokladá sa, že tieto abnormality sa podieľajú na patogenéze axonopatie pri AMN. Začiatok zápalového postihnutia je s najväčšou pravdepodobnosťou ako „second-hit“ prekrývajúci axonálnu patológiu a možnú myelínovú instabilitu, ktorá postupuje u všetkých chlapcov s X-ALD. Z neznámych dôvodov asi polovica pacientov nemá v popredí cerebrálne ťažkosti a celý život sú jediné príznaky charakteru AMN.
Cerebrálny proces sa vysvetľuje ako dôsledok imunitných reakcií, kde sa voľné časti VLCFA chovajú ako lipidové antigény a môžu byť spúšťačom imunitnej odpovede vyúsťujúcej do zápalového cerebrálneho postihnutia. Ide o agresívny, demyelinizačný proces spojený s akumuláciou CD8 cytotoxických lymfocytov, lýzou oligodendrocytov, reaktívnych astrocytov so stratou axónov. Zvyšuje sa expresivita pro--inflamačných cytokínov, tumor nekrózový faktor-α, interleukín 1,2,6,12, γ-interferón a chemikín [10–12]. Pozoruhodný poznatok v patogenéze zápalového procesu je prítomnosť apoptózy mikroglie [12].
VLCFA sa ukázali tiež ako cytotoxické pre bunky nadobličky. Nie je žiadna súvislosť medzi klinickým fenotypom ochorenia a typom DNA mutácie ABCD1 génu, preto sa v patogenéze predpokladá pôsobenie modifikátora génu [1].
Terapia X-ALD
Pre ochorenie nie je dostupná žiadna kauzálna liečba, základom je symptomatická terapia. U pacientov s adrenálnou nedostatočnosťou je hormonálna substitúcia povinná a život zachraňujúca. V prípade testikulárnej insuficiencie u AMN je namieste liečba testosterónom. Dospelí pacienti s cerebrálnou formou ochorenia vyžadujú psychiatrickú starostlivosť.
Z dlhodobého pohľadu je významná snaha znížiť hladiny VLCFA.
Redukcia VLCFA diétou s per os príjmom do 3 mg na deň bola bez efektu [13]. Úspech sa dosiahol znížením endogénnej syntézy VLCFA zmesou glycerol trioleatu a glycerol trierukátu v pomere 4 : 1 nazvanou Lorenzov olej. Podávanie zmesi normalizuje hladinu VLCFA, avšak zásadne nemení klinický obraz rozvinutého ochorenia, keďže nereparuje už vzniknuté poškodenie. Podľa štúdie Mosera et al z roku 2005 je navrhutá liečba Lorenzovým olejom u asymptomatických pacientov na oddialenie nástupu neurologického postihnutia a zmiernenie priebehu [14].
Transplantácia kostnej drene (BMT) je indikovaná pre pacientov s X-cerebrálnou ALD detského a adolescentného veku v počiatočnom štádiu s IQ nad 80. Na MR má byť nález cerebrálnej demyelinizácia s prítomnosťou enhancujúceho okraja lézie. „Loes skóre“ navrhuje klasifikáciu ložísk zvýšeného signálu v T2V obraze v rozpätí 34 bodov. Pre indikáciu transplantácie má dosiahnuť MR nález hodnotu troch a viac [15]. Keď je neurologické postihnutie pokročilé, liečba transplantáciou už nie je účinná, naopak môže spôsobiť akceleráciu príznakov. Zákrok je závažný, a preto nie je odporúčaný ani pre pacientov v asymptomatickom štádiu [1].
Iné liečebné postupy
- a) Pokus o redukciu VLCFA liečbou 4-fenylbutyrátom a lovastatínom na princípe zvýšenej expresie proteínu ALDR (vysoko homológny proteín s ALDP transportným proteínom) neboli efektívne.
- b) Liečba cerebrálnej formy imunosupresívami v úvahe ovplyvniť zápalovú zložku procesu (napr. vysoko dávkovaný cyklofosfamid, kortikoidy, talidomid, venózne podávanie imunoglobulínov) nepriniesli očakávané zlepšenie.
- c) Možnosťou sú látky s antioxidačným efektom, ktorý je pozorovaný u valproátu, mechanizmom inhibície HDAC (histón deacetyláza) a môže mať význam pre pacientov s X-AMN.
- d) Pri X-AMN by sa mohli zvažovať trofické faktory IGF 1 (rastový faktor podobný inzulínu) a NT-3 (neurotrofín-3), ktoré zabezpečujú podporu oligodendroglie a axonálnu údržbu.
- e) Sľubnou je perspektíva génovej terapie pomocou autológnej transplantácie s geneticky opravenými vlastnými kmeňovými bunkami a tiež transplantácia oligodendrocytov, ako i pluripotentných kmeňových buniek na reparovanie poškodených tkanív [1,16–19].
Pacienti a metóda
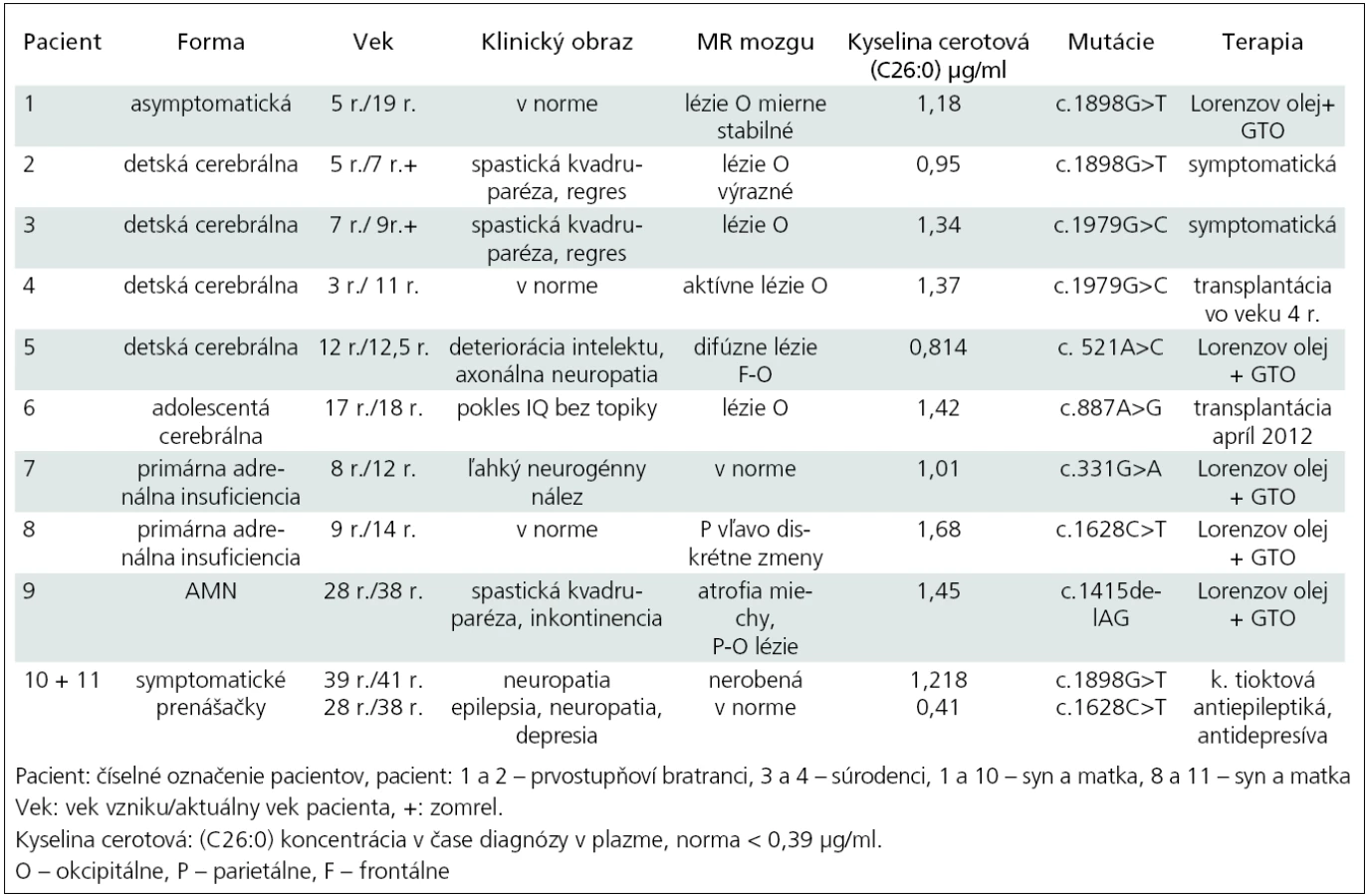
Od roku 1998 do 2011 sme na Klinike detskej neurológie v Bratislave sledovali celkom deväť pacientov s diagnózou adrenoleukodystrofie a dve nosičky (tab. 2). V súbore sú štyri detské cerebrálne formy (pacienti 2–5), najmladší chlapec bol zachytený vo veku tri roky, najstarší vo veku 12 rokov, priemerný vek na začiatku ochorenia bol šesť rokov. Pacient 1 je doteraz asymptomatický, bol zachytený po stanovení diagnózy pacienta 2 (prvostupňoví bratranci). Pacient 2 zomrel do dvoch rokov od stanovenia diagnózy. Pacientka 10 je matka pacienta 1. Z rodinnej anamnézy sme sa dozvedeli, že otec mal od 35 roku veku problémy s chôdzou a bol vedený ako progresívna forma sklerózy multiplex. Predpokladáme, že išlo o X-AMN. Pacienti 3 a 4 sú súrodenci, prvý z nich zomrel, druhý bol vo veku štyroch rokov úspešne transplantovaný. Pacient 5 je chlapec bez topickej symptomatológie so závažnými psychickými poruchami a deterioráciou intelektu. Pacient 6 je s adolescentnou cerebrálnou formou, predtým sledovaný niekoľko rokov pre m. Addison a pre zhoršenie prospechu sa podrobil MR mozgu. Pacient 7 má tiež m. Addison, čo bolo dôvodom na vyšetrenie VLCFA. Pacient 8 je rovnako s primárnou adrenálnou insuficienciou a pacientka 11 je jeho matka. Pacient 9 je s adrenomyeloneuropatiou, začiatok ťažkostí bol v 28. roku života a od 34 roku je liečený ako m. Addison s opakovanými krízami.
Všetci pacienti a nosička 11 mali MR mozgu, ktorá sa opakovala podľa stavu do desiateho roka jedenkrát za šesť až 12 mesiacov a po desiatom roku raz za rok.
Všetci mali vyšetrené VLCFA a DNA analýzu ABCD1 gén. Vyšetrovaní boli tiež príbuzní mužského pohlavia a bola uskutočnená identifikácia heterozygotiek. Pacienti 1, 5, 7, 8, 9 mali liečbu Lorenzovým olejom v dávke 0,75/kg/deň podľa tolerancie a GTO olejom pri príprave stravy. Hodnota VLCFA sa kontrolovala raz ročne. Transplantáciu podstúpili pacienti 4 a 6.
Výsledky
U všetkých pacientov boli hodnoty parametrov VLCFA v plazme: C26:0 (kyselina cerotová), prvý indexový parameter – pomer C26:0/C22:0 a druhý indexový parameter – pomer C24:0/C22:0 patologicky zvýšené. Pre prehľadnosť uvádzame v tab. 2 len údaj pre hodnotu C26:0.
V našom súbore je asymptomatický pacient 1 so stabilnými léziami na MR mozgu okcipitálne obojstranne bez známok aktivity procesu. EMG vyšetrenie dolných končatín je bez patologických zmien. Je sledovaný endokrinológom s normálnym hormonálnym profilom, bez substitúcie kortizolom. Od piateho roku života je liečený Lorenzovým olejom až doteraz. Jeho matka, pacientka 10 má parestézie nôh od 39. roku a axonálne demyelinizačnú polyneuropatiu potvrdenú EMG vyšetrením. Pacient 2 mal typické okcipitálne lézie a zomrel do dvoch rokov od začiatku ochorenia. Všetci mali identické mutácie.
Pacient 3 bol s plne rozvinutou cerebrálnou formou, s okcipitálnymi léziami a progresívnymi priebehom. Pacient 4, jeho brat, sa zachytil vo včasnom štádiu ochorenia, mal identickú mutáciou ABCD1 génu. Vo veku štyri roky bol bez neurologického deficitu. Pre prítomnosť aktívnych lézii na MR mozgu (Loes skóre 4) bol transplantovaný súrodeneckou transplantáciou pupočníkovej krvi od zdravej sestry, ktorá neniesla žiadnu mutáciu. Transplantácia prebehla úspešne, chlapec – teraz už 11-ročný – je bez ťažkostí, prerušil však kontakt s našou kliniku a informácie o zdravotnom stave sú sprostredkované z transplantačného centra [20].
Pacient 5 s cerebrálnou formou mal výrazné MR zmeny mozgu, ktoré sú zriedkavé pre X-ALD, keďže sú splývavé, difúzne frontálne aj parieto-okcipitálne. Chlapec nemá topickú neurologickú symptomatológiu, v popredí je deteriorácia intelektu. Endokrinologickým vyšetrením boli potvrdené známky adrenálnej insuficiencie a je na substitučnej liečbe kortizolom. V čase diagnózy už nespĺňal kritéria na transplantáciu kostnej drene pre Loes skóre vyššie ako 10. Pacient 6 s adolescentnou cerebrálnou formou bol sledovaný ako m. Addison. Pre prítomnosť aktívnych MR zmien a vek sme nález konzultovali s profesorom Raymondom z Kennedyho inštitútu v Baltimore (USA). Bola doporučená urgentná transplantácia, ktorá prebehla v apríli 2012 ako nepríbuzenská alogénna transplantácia kostnej drene (HLA 10/10) doteraz s dobrým priebehom. Pacient 7 (v EMG ľahká neurogénna lézia) a 8 majú primárnu adrenálnu insuficienciu a sú na trvalej substitučnej liečbe kortizolom. MR mozgu je bez alebo len s minimálnymi zmenami a nie je aktivita v MR obraze. Liečbu Lorenzovým olejom berie už len pacient 8, prvý z nich po troch rokoch liečbu odmietol. Posledný pacient 9 má adrenálnu isuficienciu na substitúcii, v klinickom obraze je spastická paraparéza s inkontinenciou a na MR obraz atrofie miechy. Päť rokov užíva Lorenzov olej. Pacientka 11 je matka pacienta 8. Pani má od 29. roku života parciálne komplexné záchvaty s generalizáciou, depresívny syndróm a nález axonálne-demyelinizačnej neuropatie na dolných končatinách. Obaja majú identické mutácie.
Všetci pacienti z nášho súboru mali vyšetrený ABCD1 gén (tab. 2) [21].
Pri podávaní Lorenzovho oleja 0,75 ml/kg/deň klesali hodnoty parametrov VLCFA a počas priebežného sledovania hodnoty kolísali. V tab. 3. uvádzame prehľad hodnôt za prvé dva roky pre všetky parametre (u pacienta 8 v prvom roku podávania prvý indexový parameter VLCFA dosiahol hornú hranicu normy a u pacienta 9 podobne druhý indexový parameter). Myslímesi, že na kolísanie hladín môže mať podiel aj vynechanie alebo redukcia dávky. Dôvody boli hlavne ťažšia spolupráca detí a v prípade pacienta 9 opakované adrenálne krízy s vracaním. Takmer všetci chlapci v začiatku podávania horšie tolerovali chuť a objem prípravku a pacient 7 liečbu neskôr odmietol. Najlepšie znáša liečbu dospelý pacient 9. Najčastejší nežiaduci účinok bol trombocytopénia bez hematologickej komplikácie.

Diskusia a záver
Podľa dostupných informácií z pracoviska, kde sa stanovujú parametre VLCFA a robí sa DNA analýza ABCD1 génu (jediné na Slovensku), bolo v období od 1998 až 2011 zachytených 20 pacientov a 30 nosičiek. Na našom pracovisku sledujeme 11 z nich. Ochorenie uvádzame v detailnejšom popise klinického obrazu. Zvlášť dospelí pacienti s X-AMN môžu byť niekedy vedení pod diagnózou chronického demyelinizujúceho ochorenia.
Myslíme si, že je významná cielená edukácia endokrinológov, kde je ďalšia možnosť zachytiť prípady aj bez neurologickej symptomatológie. Všetci pacienti mužského pohlavia s idiopatickou Addisonovou chorobou by mali mať stanovenú hladinu VLCFA na vylúčenie X-ALD. Pacienti s patologickými hodnotami potrebujú ďalšie vyšetrenia (DNA analýzu ABCD1 génu, neurologické vyšetrenie, MR, EMG) a podľa nálezov sledovanie a adekvátnu liečbu. Dôležitá je tiež laboratórna analýza rodinných príslušníkov pacienta pre možný výskyt asymptomatických homozygotov a nosičiek.
Chlapci s potvrdenou X-ALD do veku 10 rokov podliehajú psychologickému a neurologickému sledovaniu s pravidelnými kontrolami MR mozgu (podľa nálezu každých šesť mesiacov) [22]. Liečba transplantovaním kostnej drene má presné pravidlá a vykonáva sa v začiatočnom štádiu ochorenia. Podľa nových biochemických výskumov je skríningová metóda na zachytenie frakcie fosfatidil cholínu technikou hmotnostnej spektrometrie zo suchej kvapky, čo umožňuje selektovať postihnutých jedincov už v novorodeneckom veku [6]. Prácou sme chceli sumarizovať súčasný stav poznatkov a uviesť náš súbor pacientov s klinickým obrazom, vývojom a liečbou za obdobie rokov 1998 až 2011.
Zoznam použitých skratiek
- ALDP – AdrenoLeukoDystrophy Protein
- ACTH – adrenokortikotropný hormón
- ADHD – Attention Deficit Hyperactivity Disorders
- BMT – Bone Marrow Transplantation
- EMG – elektromyografické vyšetrenie
- FLAIR – FLuid-Attenuated Inversion Recovery
- MR – magnetická rezonancia
- SM – skleróza multiplex
- T2V – T2 vážený obraz na magnetickej rezonancii
- VLCFA – Very Long Chain Fatty Acids
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr. Miriam Kolníková
Klinika detskej neurológie
LF UK a DFNsP
Limbova 1
833 40 Bratislava
e-mail: kolnikova@dfnsp.sk
Prijato k recenzii: 16. 4. 2012
Prijato do tlače: 13. 9. 2012
Sources
1. Raymond GV. X-Linked Adrenoleukdystrophy. In: Raymond GV, Eichler F, Fatemi A et al (eds). Leukodystrophies. London: Mac Keith Press 2011: 75–89.
2. Moser HW, Raymond GV, Dubey P. Adrenoleukodystrophy: new approaches to a neurodegenerative disease. JAMA 2005; 294(24): 3131–3134.
3. van Geel BM, Bezman L, Loes DJ, Moser HW, Raymond GV. Evolution of phenotypes in adult male patients with X-linked adrenoleukodystrophy. Ann Neurol 2001; 49(2): 186–194.
4. Chandoga J, Petrovič R, Futas J, Ďurovčíková D, Štofko J, Jančo S et al. X-viazaná adrenoleukodystrofia – najčastejšia dedičná metabolická porucha peroxizómov. Neurol Prax 2006; 7(2): 90–95.
5. Moser AB, Kreiter N, Bezman L, Lu S, Raymond GV, Naidu S et al. Plasma very long chain fatty acids in 3,000 peroxisome disease patients and 29,000 controls. Ann Neurol 1999; 45(1): 100–110.
6. Hubbard WC, Moser AB, Tortorelli S, Liu A, Jones D, Moser H. Combined liquid chromatography tandem mass spectrometry as an analytical method for high throughput screening for X-linked adrenoleukodystrophy and other peroxisomal disorders: preliminary findings. Mol Genet Metab 2006; 89(1–2): 185–187.
7. Forss-Petter S, Werner H, Berger J, Lassmann H, Molzer B, Schwab MH et al. Targeted inactivation of the X-linked adrenoleukodystrophy gene in mice. J Neurosci Res 1997; 50(5): 829–843.
8. Kobayashi T, Shinnoh N, Kondo A, Yamada T. Adrenoleukodystrophy protein-deficient mice represent abnormality of very long chain fatty acid metabolism. Biochem Biophys Res Commun 1997; 232(3): 631–636.
9. Lu JF, Lawler AM, Watkins PA, Powers JM, Moser AB, Moser HW et al. A mouse model for X-linked adrenoleukodystrophy. Proc Natl Acad Sci USA 1997; 94(17): 9366–9371.
10. Paintlia AS, Gilg AG, Khan M, Singh AK, Barbosa E, Singl I. Correlation of very long chain fatty acid accumulation and inflammatory disease progression in childhood X-ALD: implications for potential therapies. Neurobiol Dis 2003; 14(3): 425–439.
11. Powers JM, Pei Z, Heinzer AK, Deering R, Moser AB, Moser HW et al. Adreno-leukodystrophy: oxidative stress of mice and men. J Neuropathol Exp Neurol 2005; 64(12): 1067–1079.
12. Eichler FS, Ren JQ, Cossoy M, Rietsch AM, Nagpal S, Moser AB et al. Is microglial apoptosis an early pathogenic change in cerebral X-linked adrenoleukodystrophy? Ann Neurol 2008; 63(6): 729–742.
13. Rizzo WB, Leshner RT, Odone A, Dammann AL, Craft DA, Jensen ME et al. Dietary erucic acid therapy for X-linked adrenoleukodystrophy. Neurology 1989; 39(11): 1415–1422.
14. Moser HW, Raymond GV, Lu SE, Muenz LR, Moser AB, Xu J et al. Follow-up of 89 asymptomatic patients with adrenoleukodystrophy treated with Lorenzo’s oil. Arch Neurol 2005; 62(7): 1073–1080.
15. Seidl Z, Vaněčková M, Vítek T, Kron M, Dvořáková L, Zeman J. X-adrenoleukodystrofie-hodnocení lézí mozku modalitou magnetické rezonance pomocí „Loes score“. Cesk Radiol 2007; 61(3): 275–278.
16. Cavaletti G. Current status and future prospective of immunointervantion in multiple sclerosis. Curr Med Chem 2006; 13(19): 2329–2343.
17. Linsen L, Somers V, Stinissen P. Immunoregulation of autoimmunity by natural killer T cells. Hum Immunol 2005; 66(12): 1193–1202.
18. Berger J, Pujol A, Auborg P, Forss-Petter S. Current and future pharmacological treatment strategies in X-linked adrenoleukodystrophy. Brain Pathol 2010; 20(4): 845–856.
19. Cartier N, Hacein-Bey-Abina S, Bartholomae CC, Veres G, Schmidt M, Kutschera I et al. Hematopoetic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science 2009; 326(5954): 818–823.
20. Horáková J, Lukáč J, Šufliarska S, Boďová I, Mydliar M, Sýkora P et al. Transplantácia pupočníkovej krvi u 4-ročného chlapca s adrenoleukodystrofiou viazanou na chromozóm X. Cesk Pediatr 2005; 4: 219–223.
21. Dvorakova L, Storkanova G, Unterrainer G, Hujova J, Kmoch S, Zeman J et al. Eight novel ABCD1 gene mutations and three polymorphisms in patients with X-linked adrenoleukodystrophy: the first polymorphism causing an amino acid exchange. Hum Mutat 2001; 18(1): 52–60.
22. van der Knaap MS. X-linked adrenoleukodystrophy. In: van der Knaap MS, Valk J (eds). Magnetic Resonance of Myelination and Myelin Disorders. 3rd ed. Berlin Heidelberg New York: Springer 2005: 176–190.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2013 Issue 2
Most read in this issue
- Creutzfeldtova-Jakobova choroba
- Spinocerebelární ataxie typ 7 (SCA7) – kazuistika
- Lymeská borelióza jako příčina bilaterální neuroretinitidy s výraznou jednostrannou hvězdicovitou makulopatií u osmileté dívky
- Elektrofyziologické vyšetření pánevního dna