
Creutzfeldtova-Jakobova choroba
Creutzfeldt-Jacob disease
Creutzfeldt-Jacob disease (CJD) is a prion disease. Prion diseases are fatal neurodegenerative conditions affecting humans and a wide variety of animals. In prion diseases, normal, cellular protein (PrPC) is converted into insoluble, protease-resistant scrapie prion protein (PrPSC). PrP is encoded by the prion protein gene (PRNP) localised on the short arm of chromosome 20. There are several forms of CJD, sporadic CJD (sCJD), seen in about 85% of patients, being the most common one. Some cases are genetic (gCJD) that occur as a consequence of various mutations (including point mutations, insertions and deletions); when the mutation is present in more than two members of a family, the disease is called familial (fCJD). Remaining two forms, i.e. iatrogenic CJD (iCJD) and variant CJD (vCJD), are acquired. iCJD (now called accidentally transmitted) is acquired through contaminated transplants and instruments. vCJD is a novel form of human prion disease first reported in the United Kingdom in 1996. It is likely that bovine prions from „mad cows“, affected with bovine spongiform encefalopathy (BSE), were passed to humans through consumption of beef products from affected animals.
Key words:
Creutzfeldt-Jacob disease – prion protein – 14-3-3 protein – electroencephalography – magnetic resonance imaging – dementia
Authors:
Z. Gdovinová
Published in:
Cesk Slov Neurol N 2013; 76/109(2): 138-154
Category:
Minimonography
Overview
Creutzfeldtova-Jakobova choroba (CJch) patrí medzi prionové ochorenia, smrteľné neurodegeneratívne ochorenia, postihujúce tak ľudí, ako aj zvieratá. Sú charakterizované konverziou bunkového prionového proteínu PrPC na abnormálnu, nerozpustnú a čiastočne proteázo-rezistentnú izoformu nazývanú scrapie priónový proteín (PrPSC). Gén pre syntézu celulárneho PrP človeka (PRNP) je lokalizovaný na krátkom ramienku chromozómu 20. Rozlišujeme niekoľko foriem CJch, najčastejšou formou CJch je sporadická Creutzfeldtova-Jakobova choroba (sCJch), ktorá sa vyskytuje asi u 85 % pacientov, zriedkavejšie sa vyskytuje genetická forma (gCJch), ktorá vzniká na základe rôznych typov mutácií (bodové mutácie, inzercie, delécie), pri výskyte u viac ako dvoch členov rodiny sa označuje ako familiárna forma (fCJch). Iatrogénna (iCJch), v súčasnosti označovaná ako náhodne prenesená (accidentally transmitted), vznikla prenosom z človeka na človeka kontaminovanými transplantátmi alebo nástrojmi. Nový variant (vCJch) bol prvýkrát popísaný vo Veľkej Británii v roku 1996 a vznikol s vysokou pravdepodobnosťou prenosom z hovädzieho dobytka pri „chorobe šialených kráv“ – bovinnej spongiformnej encefalopatii (BSE).
Kľúčové slová:
Creutzfeldtova-Jakobova choroba – prionový proteín – protein 14-3-3 – elektroencefalografia – magnetická rezonancia – demencia
História
Creutzfeldtova-Jakobova choroba patrí medzi humánne prionové choroby. Prvýkrát bolo ochorenie spomenuté v roku 1920, kedy Hans Gerhard Creutzfeldt popísal prípad 22-ročnej ženy, ktorá zomrela na progresívnu cerebrálnu dysfunkciu. O rok neskôr Alfons Maria Jakob popísal ďalších 5 pacientov. Názov Creutzfeldtova-Jakobova choroba (CJch) vznikol v roku 1922, ale prešiel aj obdobím rôznych synoným [1]. V roku 1968 nositeľ Nobelovej ceny Carleton Daniel Gajdušek úspešne preniesol kuru a Creutzfeldtovu-Jakobovu chorobu človeka na laboratórne zviera, čím sa vytvorila možnosť dôkazu CJch experimentálnym prenosom nákazy [2,3]. Spočiatku sa predpokladalo, že etiologickým agensom ochorenia sú nekonvenčné vírusy, názov prionové ochorenia vznikol po objavení prionu ako pôvodcu ochorenia v r. 1982 Prusinerom, za čo v roku 1997 získal Stanley B. Prusiner Nobelovu cenu za fyziológiu a medicínu [4].
Dnes rozlišujeme niekoľko foriem CJch, najčastejšou formou je sporadická Creutzfeldtova-Jakobova choroba (sCJch), ktorá sa vyskytuje asi u 85 % pacientov, zriedkavejšie sa vyskytuje genetická forma (gCJch), ktorá vzniká na základe rôznych typov mutácií (bodové mutácie, inzercie, delécie), pri výskyte u viac ako 2 členov rodiny sa označuje ako familiárna forma (fCJch). Iatrogénna (iCJch) vznikla prenosom z človeka na človeka kontaminovanými transplantátmi alebo nástrojmi. Nový variant (vCJch) bol prvýkrát popísaný vo Veľkej Británii v roku 1996 a vznikol s vysokou pravdepodobnosťou prenosom z hovädzieho dobytka pri „chorobe šialených kráv“ – bovinnej spongiformnej encefalopatii (BSE).
V roku 1993 zahájila Európska únia spoločný program dohľadu nad výskytom „Creutzfeldt-Jakob disease“ (CJch) – surveillance CJch – EUROCJch, k pôvodným ôsmym štátom, medzi ktorými bolo aj Slovensko, neskôr pribudli nielen ďalšie štáty Európy, ale aj Austrália, Kanada a USA. Spoločný postup umožnil získať štatisticky hodnotiteľné súbory, údaje o priemernom ročnom výskyte CJch a jej foriem. Slovensko, kde sa povinné hlásenie CJch datuje od roku 1983, charakterizuje unikátny, až 69% podiel genetickej formy s mutáciou E200K (gCJchE200K), s koncentráciou hlavne na Orave, Kysuciach a okolo Rožňavy [5]. Na našom pracovisku boli hospitalizovaní 4 pacienti s mutáciou prionového génu E200K v kodóne 200, dvaja z Košíc a dvaja z Trebišova.
Patofyziológia
Prionové ochorenia
Prionové ochorenia sú smrteľné neurodegeneratívne ochorenia postihujúce tak ľudí, ako aj zvieratá. Sú charakterizované konverziou bunkového prionového proteínu PrPC na abnormálnu, nerozpustnú a čiastočne proteázo-rezistentnú izoformu nazývanú scrapie priónový proteín (PrPSC). Fyziologický PrP sa nachádza na bunkovej membráne neurónov, ale aj v ďalších tkanivách, jeho presná fyziologická funkcia nie je stále presne objasnená [6]. Gén pre syntézu celulárneho PrP človeka (PRNP) je lokalizovaný na krátkom ramienku ľudského chromozómu 20. Skladá sa z dvoch exónov, pričom druhý exón obsahuje kompletný otvorený čítací rámec (ORF). Kóduje polypeptidový reťazec s 253–254 aminokyselinami, zrelý proteín po translačných úpravách pozostáva z 208 aminokyselín o molekulovej hmotnosti 35–36 kDa. Mutácie tohoto génu môžu napomáhať transformácii PrPC proteínu na jeho patologickú izoformu PrPSC. Je známych viac ako 25 takýchto mutácií. Pri konverzii normálneho PrPC na patologickú izoformu PrPSC sa počet a sekvencia aminokyselín nemení, ale dochádza ku konformačným zmenám. PrPSC sa naviaže na PrPC a nie celkom známym spôsobom ho premení na PrPSC. Dochádza k reťazovej reakcii, pri ktorej sa PrPSC hromadia v nervových bunkách, tie sa poškodzujú, dochádza k ich vakuolizácii a následnému odumretiu. Priony sa ukladajú aj v mimobunkovom priestore, kde sa zhlukujú do masy a vytvárajú tzv. amyloidové plaky. Všetky funkcie prionu u zdravého človeka nepoznáme, ale podieľa sa na synaptickom prenose vzruchu, antioxidatívnych procesoch, funkcii biologických hodín a pravdepodobne aj pri ukladaní informácii do pamäti. Rozdiely medzi oboma proteínmi sú v ich sekundárnej štruktúre, a to v pomere α-helixu a β-štruktúry. Molekula PrPC obsahuje 42 % α-helixu a len 3 % β-štruktúry, kým priestorové usporiadanie PrPSC predstavuje len 30 % α-helixu, ale až 43 % β-štruktúry, ktorá je odolnejšia voči proteolytickým enzýmom (tab. 1) [7,8]. V dôsledku rovnakej primárnej štruktúry oboch proteínov nerozoznáva imunitný systém PrPSC ako cudzí, a preto sa proti nemu netvorí imunita.
![Rozdiely medzi celulárnym priónovým proteínom (PrPC) a scrapie priónovým proteínom (PrP<sup>SC</sup>), upravené podľa [7].](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image/b046e1f5fdec53ca2626b62e8e39c73e.png)
Pre lepšie pochopenie podrobnejšej klasifikácie prionových ochorení vrátane rozdelenia CJch na 6 subtypov je potrebné vysvetlenie pojmu PrPres, ktorý zaviedol Gambetti et al [10]. Na základe štiepenia PrP proteinkinázou K vznikajú dva proteíny odlišnej hmotnosti, 21 kDa a 19kDa, označované ako typ 1 a typ 2 PrPres (PrPres – PrP rezistentný na proteázu) [11].
Okrem toho sa prionové proteíny rozlišujú aj na základe glykozylácie. Glykozylácia proteínov, tj. pripojenie cukornej zložky, patrí medzi dôležité postranslačné úpravy proteínov. V prípade PrP sú variabilne glykozylované aminokyseliny Asn180 alebo Asn196, to znamená, že môžu, ale nemusia byť k nim naviazané 1–2 oligosacharidové reťazce. Jedná sa o N väzbové oligosacharidy, ktoré zohrávajú významnú úlohu pri skladaní proteínov. Pomer neglykozylovaných, monoglykozylovaných a diglykozylovaných glykoforiem tak ovplyvňuje výslednú konformáciu a stabilitu PrP [12].
Jednotlivé humánne prionové ochorenia sú charakterizované rozdielnou konformáciou a glykozyláciou PrPres, teda rozdielnou dostupnosťou jednotlivých oblastí prionového proteinového polypeptidového reťazca pre štiepenie proteinkinázou K. Collinge et al [13] a Telling et al [14] na základe týchto poznatkov dospeli k názoru, že rozdielne konforméry a glykotypy ovplyvňujú fenotypové vlastnosti ľudských prionov.
U väčšiny ľudí s prionovým ochorením je príčina neznáma, u části pacientov ochorenie vzniká prenosom zo známeho zdroja infekcie a 10–15 % pacientov má genetickú formu ochorenia buď v dôsledku bodovej mutácie, alebo v dôsledku inzercie oktapeptidových opakovaní (OPR) v géne pre prionový proteín (PRNP). Títo majú často pozitívnu rodinnú anamnézu s autozomálne dominantným typom dedičnosti s variabilnou penetranciou. Prionové ochorenia sa vyznačujú rozsiahlou fenotypovou heterogenitou. Tri najčastejšie formy sú: Creutzfeldtova-Jakobova choroba (CJch), fatálna familiárna insomnia (FFI) a Gerstmannov-Sträusslerov-Scheinkerov (GSS) syndróm [9].
Creutzfeldtova-Jakobova choroba (CJch)
Incidencia CJch je 1–2 pacienti na 1 000 000 obyvateľov ročne, najčastejšou formou CJch je sporadická Creutzfeldtova-Jakobova choroba (sCJch), ktorá sa vyskytuje asi u 85 % pacientov [15], zriedkavejšie sa vyskytuje genetická forma (gCJch). Genetická forma vzniká v dôsledku celého radu mutácií (bodové mutácie, inzercie, delécie) v géne pre prionový protein (PRNP) (obr. 1) [16]. Zvyšok tvoria získané formy – iatrogénna CJch (iCJch) a nový variant (vJCch) [17]. Iatrogénna vznikla z kontaminovaných rastových hormónov od kadáverov, z dura mater, časť bola prenesená neurochirurgickými nástrojmi, EEG elektródami pri stereotaktických vyšetreniach a transplantáciou rohovky [17]. Nový variant vznikol veľmi pravdepodobne prenosom z hovädzieho dobytka pri „chorobe šialených kráv“ – bovinnej spongiformnej encefalopatii (BSE). Vzťah vCJch k BSE určila typizácia kmeňa priónov a experimentálny prenos na transgénnu aj konvenčnú myš [18].
![Schéma génu pre priónový proteín (PRNP) a mutácie. Schematická reprezentácia génu PRNP s najvýznamnejšími polymorfizmami a mutáciami. Všetky mutácie sú asociované s CJch okrem tých, ktoré sú znázornené boldom (GSS – Gerstmannov-Sträusslerov-Scheinkerov syndróm), plným rámčekom (FFI – fatálna familiárna insomnia alebo CJch, v závislosti od genotypu v kodóne 129) a bodkovaným rámčekom (fenotyp CJch s variabilnou patológiou).
Upravené podľa [16].](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image/4f10dba5093e52c6b19e9ea3fcceef40.png)
Sporadická Creutzfeldtova-Jakobova choroba
Epidemiológia
Incidencia sCJch je 1–2 pacienti/1 mil. obyvateľov. Nie je rozdiel vo výskyte medzi pohlaviami, priemerný vek začiatku ochorenia je okolo 65 rokov. V minulosti sa za rizikový faktor sCJch považovala konzumácia mozgu a očí oviec postihnutých klusavkou (scrapie), avšak výskyt ochorenia celosvetovo, aj v oblastiach, kde sa scrapie nevyskytuje alebo má len veľmi nízku incidenciu, svedčí proti tejto teórii. Nedávne objavenie sa atypických foriem scrapie a bovinnej spongiformnej encefalopatie (BSE) obnovili úvahy o medzidruhovom prenose (cross-species transmision) na ľudí. Tieto úvahy posilnili aj výsledky experimentálneho transgénneho prenosu L-typu BSE (BASE) humánnym PRNP na primáty [19] a na myši [20,21]. Ďalším zvažovaným rizikovým faktorom bola predchádzajúca operácia pacienta kontaminovanými inštrumentami, avšak podľa veľkej štúdie realizovanej vo Veľkej Británii je málo pravdepodobné, aby veľká časť sCJch bola zapríčiená operáciou [22].
Genetika
Kodón 129 PRNP génu obsahuje polymorfizmus (ATG/GTG), ktorý kóduje metionín (M) alebo valín (V). Samotný polymorfizmus nie je patogenetický, ale homozygotná varianta MM zvyšuje riziko sCJch (približne 72 % pre MM, 17 % pre VV) [21], kým heterozygotný genotyp má akoby protektívny charakter. Jeho frekvencia u sJCch je nižšia (17 %) ako v bežnej populácii (51 %). Genotyp v kodóne 129 spolu s typom 1 alebo 2 PrPres majú výrazný vplyv aj na klinickopatologický fenotyp [23].
Diagnostika
Diagnostika sCJch je založená na klinickom obraze, priebehu ochorenia a výsledkoch pomocných vyšetrení – EEG, stanovenie proteínu 14-3-3 v likvore a vyšetrenie mozgu magnetickou rezonanciou (FLAIR MR a DWI MR). Pre definitívnu diagnózu je potrebné potvrdenie neuropatologicky/imunocytochemicky. WHO boli v roku 1998 v Ženeve stanovené odporučenia pre diagnostiku sCJch, v roku 2003 bol vypracovaný Manuál WHO pre diagnostiku sCJch (dostupný na http://whqlibdoc.who.int/publications/2003/9241545887.pdf) a tieto dokumenty boli neskôr MR-CJch konzorciom doplnené o nálezy pri magnetickej rezonancii mozgu a v auguste 2010 boli centrom pre kontrolu a prevenciu ochorení (http://www.cdc.gov) publikované aktuálne platné kritériá (tab. 2) [24–26].
![Diagnostické kritériá pre sporadickú CJch – definitívna, pravdepodobná, možná podľa WHO [24,25] a MR-CJch konsorcia [26].](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image/fe38d99a29ddc065f26b70a3daaa9462.png)
Klinický obraz
K hlavným klinickým príznakom sCJch patria kognitívny deficit, ataxia, zrakové poruchy, ktoré sú prítomné buď samostatne, alebo v kombinácii. U niektorých pacientov je prítomný myoklonus, je častejší v neskorých štádiách ochorenia, ale je zriedkavý. Postupne sa pridružuje mnohopočetné postihnutie CNS s globálnou kognitívnou dysfunkciou, ataxiou, rigiditou, inkontinenciou, postupnou stratou nezávislosti a pripútaním na lôžko. V terminálnom štádiu sa často pridružuje dysfágia, ktorá býva príčinou aspiračnej pneumónie ako príčiny smrti. Priemerné trvanie ochorenia je 4,5–7,4 mesiaca. Aj v dĺžke prežívania je rozdiel medzi pacientami. Asi 10 % prežíva viac ako rok, 5 % viac ako 2 roky [24,27]. Najkratšie popísané prežívanie bolo 2 týždne.
Subtypy sporadickej Creutzfeldtovej-Jakobovej choroby
Rozlišujeme 6 subtypov sporadickej CJch. Na molekulárnej úrovni korelujú s genotypom v kodóne 129 (MM, MV, VV) a s fyzikálnochemickými vlastnosťami PrPres (typ 1 alebo typ 2). Niekedy sa môžu vyskytovať 2 subtypy súčasne.
Najčastejší typ sCJch, ktorý sa v čistej forme (MM1) vyskytuje asi v 40 % a v zmiešanej forme (MM1 + MV1) asi v 70 %, je u homozygotov alebo heterozygotov pre metionín v kodóne 129, ktorí majú PrPres typ 1, teda MM1 a MV1 [27–29], ďalej sú to subtypy VV2, MV2, MM2 a VV1. Typické klinické príznaky a nálezy pomocných vyšetrení sa v jednotlivých prácach mierne odlišujú, čo môže byť dané aj malým počtom pacientov v jednotlivých súboroch. Najtypickejšie nálezy na základe spracovania viacerých štúdií sú uvedené v tab. 3 [10,13,26,27,30,31]. Cieľom uvedenia klinického obrazu jednotlivých subtypov sCJch je poukázať na možný rozdielny priebeh ochorenia u jednotlivých pacientov. Z každodennej klinickej praxe máme skúsenosť, že pri menej typickom priebehu sa na ochorenie nemyslí ani v pokročilejších štádiách.
![Najtypickejšie klinické, EEG, likvorové a MR nálezy u jednotlivých subtypov sporadickej Creutzfeldtovej-Jakobovej choroby (sCJch) na základe spracovania viacerých štúdií [10,13,26,27,30,31].](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image/16f2daf7b3d9020eb84c48f5da97c3a4.png)
Viaceré štúdie ukázali, že u jedného pacienta sa môžu vyskytovať PrPres typ 1 a PrPres typ 2 samostatne aj súčasne. Súčasný výskyt modifikuje vyššie popísané klinické obrazy [32].
Z praktického hľadiska a pre rýchlejšiu orientáciu Puoti G et al [31] uvádzajú rozdelenie prionových ochorení na 3 hlavné skupiny, z ktorých sa sporadickej CJch týkajú: kognitívne subtypy: sCJch MM1 a sCJch MV1, sCJch MM2 a sCJch VV1 a subtypy s dominujúcou ataxiou: sCJch VV2 a sCJch MV2. Tretím subtypom sú sporadické prionové ochorenia s non-CJch fenotypmi, kam zaraďujú sporadickú fatálnu insomniu a prionopatiu s variabilnou senzitivitou na štiepenie proteázou – Variably Protease-Sensitive Prionopathy (VPSPr).
Kognitívne subtypy
V tejto skupine sú najčastejšie subtypy sCJch MM1 a sCJch MV1, ktoré spolu predstavujú 55–70 % sporadických prionových ochorení [27,33]. Ochorenie začína najčastejšie vo veku 50–70 rokov rýchle progredujúcim kognitívnym deficitom, zmätenosťou, príležitostne môžu byť sprevádzané kortikálnou zrakovou poruchou, ataxiou a spontánnym alebo provokovaným myoklonom. Neurologickým príznakom často predchádzajú mierne psychické príznaky ako únava, úzkosť, zmeny nálady a poruchy koncentrácie. Zriedkavo sa môžu v úvode ochorenia vyskytovať extrapyramídové alebo mozočkové príznaky, ale neurologické vyšetrenie môže byť v úvode ochorenia aj negatívne, neurologické príznaky môžu byť aj unilaterálne. Progresia ochorenia je rýchla, niekedy v priebehu niekoľkých dní, čo spolu s klinickým obrazom pomerne rýchlo vzbudzuje podozrenie na prionové ochorenie a vedie k realizácii MR mozgu, vyšetrenia likvoru a EEG a spolu s týmito vyšetreniami k stanoveniu diagnózy sCJch. V priebehu niekoľkých týždňov dochádza k rozvoju závažných pyramídových a extrapyramídových príznakov, poruche vedomia a k úmrtiu v priebehu približne 4 mesiacov od začiatku ochorenia.
Subtypy sCJch MM2 a sCJch VV1 sú zriedkavejšie (2–10 %), je ich ťažšie rozlíšiť na základe klinického obrazu, u obidvoch dominuje kognitívny deficit a pacienti môžu byť niekoľko mesiacov monosymptomatickí [27,33]. U sCJch MM2 môže dominovať amnestická afázia a apraxia [31]. Aj keď celkový priebeh býva dlhší (okolo 14 mesiacov), kognitívny deficit progreduje rýchlo a v priebehu 5 mesiacov sa vyvíja ťažká demencia [31]. U väčšiny pacientov je už v tomto počiatočnom štádiu pozitívne MR mozgu, kým protein 14-3-3 v likvore (proteín, k uvolňovaniu ktorého dochádza v dôsledku deštrukcie nervového tkaniva) a tau proteín sú prítomné v likvore len u približne polovice pacientov a periodické komplexy ostrých vľn v EEG sú prítomné len u menej ako polovice pacientov so stredným postihnutím alebo v pokročilom štádiu. Ostatné neurologické príznaky sa pridružujú až po viac ako 5 mesiacoch.
U sCJch VV1 progreduje kognitívny deficit pomalšie, dominujú hlavne zmeny osobnosti. Hypertonia, ataxia a myoklonus sa vyvíjajú až po viac ako 7 mesiacoch. Typickým príznakom sCJch VV1 je relatívne nízky vek začiatku ochorenia, okolo 41 rokov veku, čo spolu s poruchami správania a pomalšou progresiou môže viesť k zámene s novým variantom CJch. V diferenciálnej diagnóze má význam MR mozgu s nálezom hyperintenzity mozgovej kôry alebo bazálnych ganglií alebo v obidvoch oblastiach, kým u vCJch je už vo včasných štádiách obojstranne prítomný príznak pulvinar thalami [27,34,35]. Pri sCJch je skoro u všetkých pacientov prítomný protein 14-3-3 v likvore, kým u vCJch len asi u 50% pacientov a rozdiel je aj v genotype, všetci doteraz diagnostikovaní pacienti s vCJch boli v kodóne 129 homozygoti pre metionín. Zásadným rozdielom je dôkaz PrPSC v tkanive z biopsie tonzíl u pacientov s vCJch [36].
Subtypy s príznakmi ataxie
Prevalencia týchto subtypov je nižšia, dominujú sCJch VV2 a sCJch MV2, predstavujú asi 1/3 pacientov so sCJch. Je pre nich, ako už názov hovorí, typický výskyt ataxie, ale bežne je prítomný aj kognitívny deficit, hlavne u sCJch MV2 [27,37]. Ataxia a kognitívny deficit môžu byť niekoľko mesiacov jedinými príznakmi ochorenia, až neskôr sa objavujú extrapyramídové príznaky a myoklonus. Ako prvé príznaky sú často prítomné zmeny nálady, závrativosť, poruchy rovnováhy. Práve pre tieto rôznorodé príznaky, neskoré objavenie sa myoklonu a zriedkavo pozitívny EEG nález sú príčinou problematickej diagnostiky sCJch u týchto pacientov [24,37]. U obidvoch subtypov je vysoko senzitívne vyšetrenie proteínu 14-3-3 a tau proteínu v likvore [37] a MR mozgu s nálezom hyperintenzity v oblasti bazálnych ganglií a thalamu (čím sa líšia od sCJch MM1, kde je thalamus postihnutý minimálne alebo vôbec) [38].
Zobrazovacie vyšetrenia
CT vyšetrenie u sCJch nie je priekazné, asi u 60–80 % pacientov je v norme, najčastejšou abnormitou je atrofia mozgovej kôry, aj to len u pacientov s dlhším trvaním ochorenia.
Z hľadiska diagnostiky má najväčší význam MR, najvyššiu senzitivitu a špecificitu má DWI MR, ktoré skoro nahradilo FLAIR sekvenciu, v praxi sa väčšinou stále realizujú obidve vyšetrenia [26,33]. MRI-CJch konzorcium vypracovalo návrh na MR kritériá pre diagnostiku sCJch a po ich zapracovaní návrh na nové kritériá pre sCJch [26,33].
MR kritériá pre diagnostiku sCJch:
Zvýšená intenzita signálu v/vo:
- viac ako troch kôrových oblastiach mozgu,
- hipokampe,
- v ktoromkoľvek z bazálnych ganglií,
- v ktoromkoľvek jadre thalamu,
- v cerebele.
Zapracované nové kritériá do odporúčaní WHO sú uvedené v tab. 2 (v časti Diagnostika). I keď MR kritériá uvádzajú hyperintenzitu v ktoromkoľvek z bazálnych ganglií, typickým nálezom sú hyperintenzity v oblasti putamen a nucleus caudatus (obr. 2, 3 a tab. 2). Hyperintenzity v kôre sú hlavne frontálne (obr. 4) a inzulárne. U malého percenta pacientov môže byť MR nález mozgu aj falošne pozitívny. 50 % falošne pozitívnych nálezov je u pacientov so zápalovými ochoreniami (encefalitídy, SREAT – Steroid-Responsive Encephalopathy Associated with autoimmune Thyroiditis) [39,40], potom u pacientov s Alzheimerovou chorobou a demenciou s Lewyho telieskami [41].
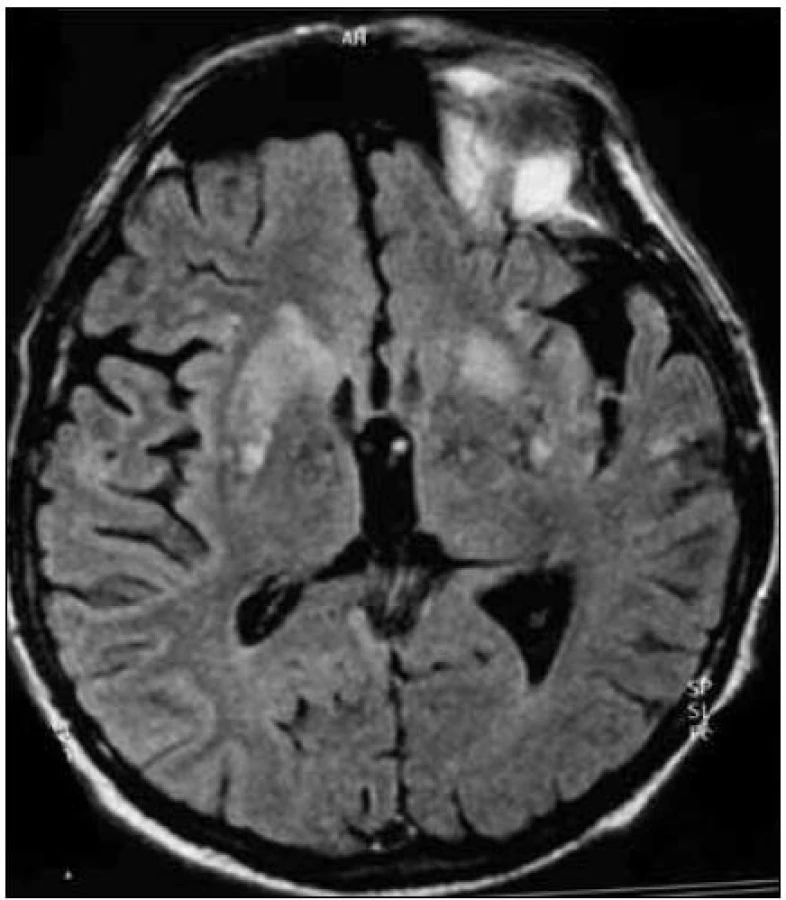
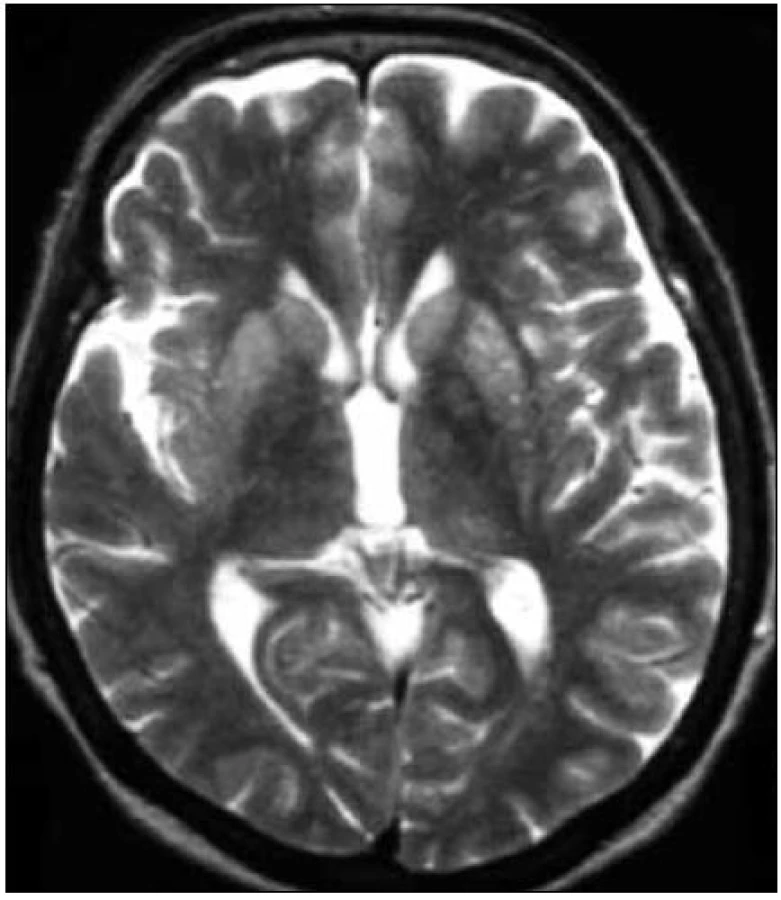
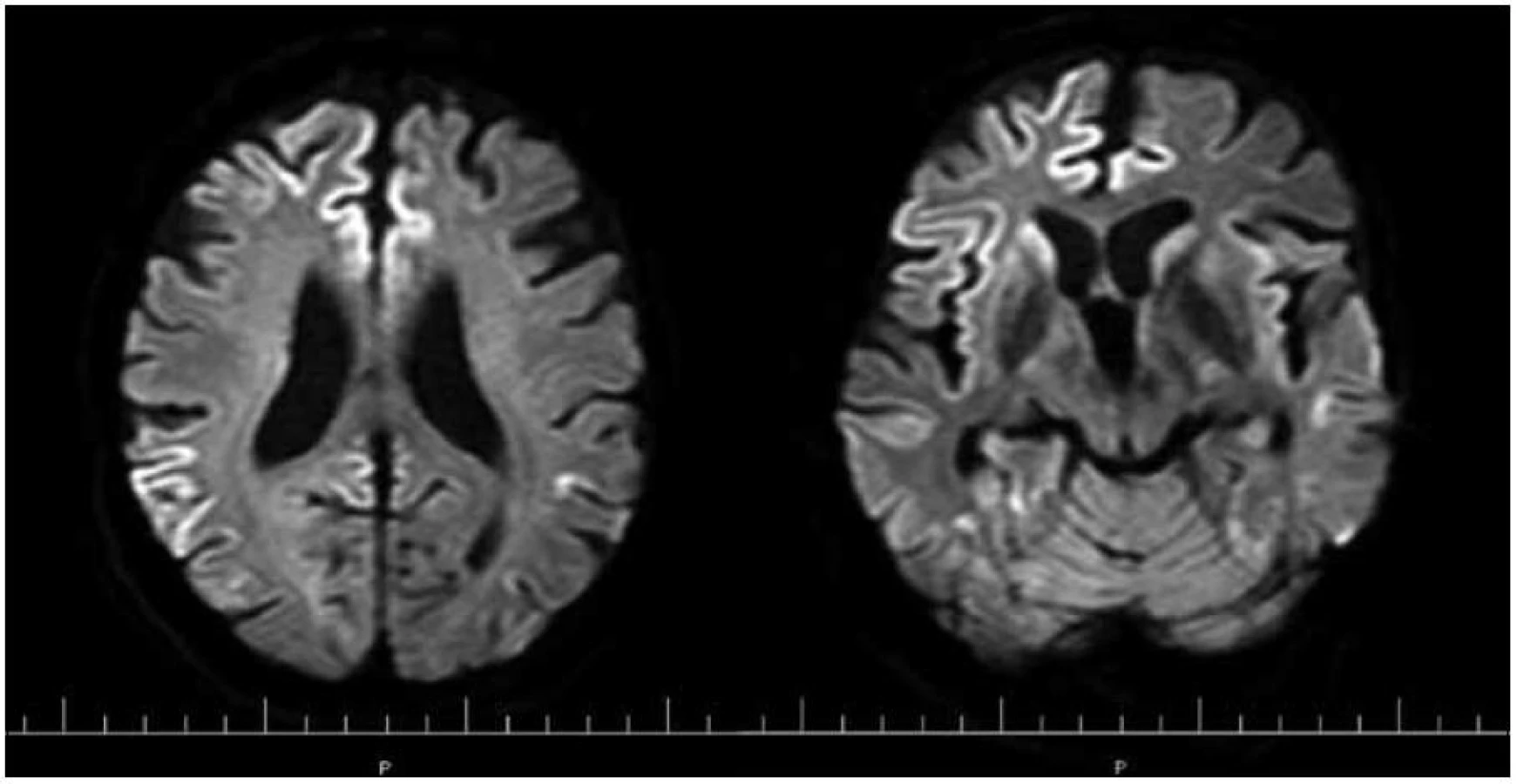
Rozdiely v MR nálezoch u jednotlivých subtypov sCJch boli popísané v časti o ich klinickom obraze.
Laboratórne testy
Likvor
Bežné laboratórne testy a vyšetrenie likvoru sú väčšinou normálne. Pre sCJch je typické zvýšenie proteínu 14-3-3 v likvore (v SR sa stanovuje v Národnom referenčnom centre pre prionové choroby, SZU Bratislava a v ČR v Národní referenční laboratoři lidských prionových ochorení při Oddělení patologie a molekulární medicíny TN v Prahe). K jeho uvolňovaniu dochádza v dôsledku deštrukcie nervového tkaniva, teda nielen u CJch, ale aj pri krvácaní do mozgu, mozgovom infarkte, tumoroch mozgu, zápalových ochoreniach CNS a inom poškodení nervového tkaniva. Dôkaz proteinu 14-3-3 v likvore teda nie je špecifický pre CJch, o jeho význame pre diagnostiku CJch sa vedú rozsiahle diskusie [42,43]. Senzitivita a špecificita tohto vyšetrenia sa udáva v obidvoch prípadoch 93 %, tento test je súčasťou protokolu pre stanovenie pravdepodobnej diagnózy sCJch. U rýchle progredujúcich neurodegeneratívnych demencií je prítomnosť β-podjednotky proteinu 14-3-3 v likvore považovaná za jeden z hlavných paraklinických diagnostických markerov sCJch [44]. Porovnateľnú presnosť má aj zvýšená hladina tau proteinu (celkového aj fosforylovaného), tento test je niekedy citlivejší ako prítomnosť proteínu 14-3-3 [45], ale v súčasnosti nie je súčasťou platných diagnostických kritérií [44,46]. Gmitterová et al [47] a Chohan et al [48] udávajú senzitivitu a špecificitu 85–97 % a 84–97 %. Mitrová [45] porovnávala senzitivitu stanovenia proteínu 14-3-3 a tau-proteínu u genetických foriem prionových ochorení a zistila podstatne vyššiu senzitivitu u gCJCh (78 %, resp. 82 %) voči FFI (9 %, resp. 0 %) a GSS (0 %, resp. 50 %). Senzitivitu stanovenia proteínu 14-3-3 a tau proteínu v likvore zvyšuje stanovenie proteínu S100b a neurón špecifickej enolázy (NSE) [49]. Niektorí autori uvádzajú, že pri negatívnom výsledku vyšetrenia likvoru by sa mal test opakovať s odstupom 2–3 týždňov [31]. Proteín 14-3-3 je najčastejšie prítomný v likvore u subtypov VV1 a VV2 sCJch, asi u 1/2 MM2 sCJch [31], u subtypu MV2 a hlavne MM2 býva prítomný menej často (tab. 3) [26,33].
Atarashi et al publikovali zavedenie nového testu v diagnostike CJch – „real-time quaking-induced conversion“ – ktorý je založený na rýchlom dôkaze minútového množstva PrPSC in vitro konverziou rekombinantného PrPC pôsobiaceho ako substrát. Senzitivita testu bola vyššia ako 83–87 % a špecificita bola 100 %, avšak bolo vyšetrených len málo pacientov, preto tieto výsledky potrebujú ešte ďalšie overenie [50].
Elektroencefalografia – EEG
Typickým EEG nálezom u pacientov s CJch je prítomnosť periodických komplexov ostrých vľn (PSWCs), ktoré musia spĺňať nasledovné kritériá [51]:
- prísne periodické cerebrálne potenciály, väčšina s trvaním medzi 100 a 600 ms a s intervalom medzi komplexami medzi 500 a 2 000 ms,
- sú akceptované aj generalizované a lateralizované komplexy,
- najmenej 5 repetitívnych intervalov s rozdielom v trvaní menej ako 500 ms vylučuje semiperiodickú aktivitu.
Tvar komplexov je bi- alebo trifázický (obr. 5) [33] (EEG záznam s prítomnosťou periodických trifázických vĺn z archívu Neurologickej kliniky). Niektorí autori uvádzajú, že PSWCs sú typické pre neskoršie štádia ochorenia, v skorších štádiách je typická prítomnosť pomalých vĺn difúzne a frontálne prítomnosť rytmickej delta aktivity [31]. Veľká prospektívna analýza však potvrdila typický EEG nález len u 144 z 219 patologicky potvrdených diagnóz sCJch [46].
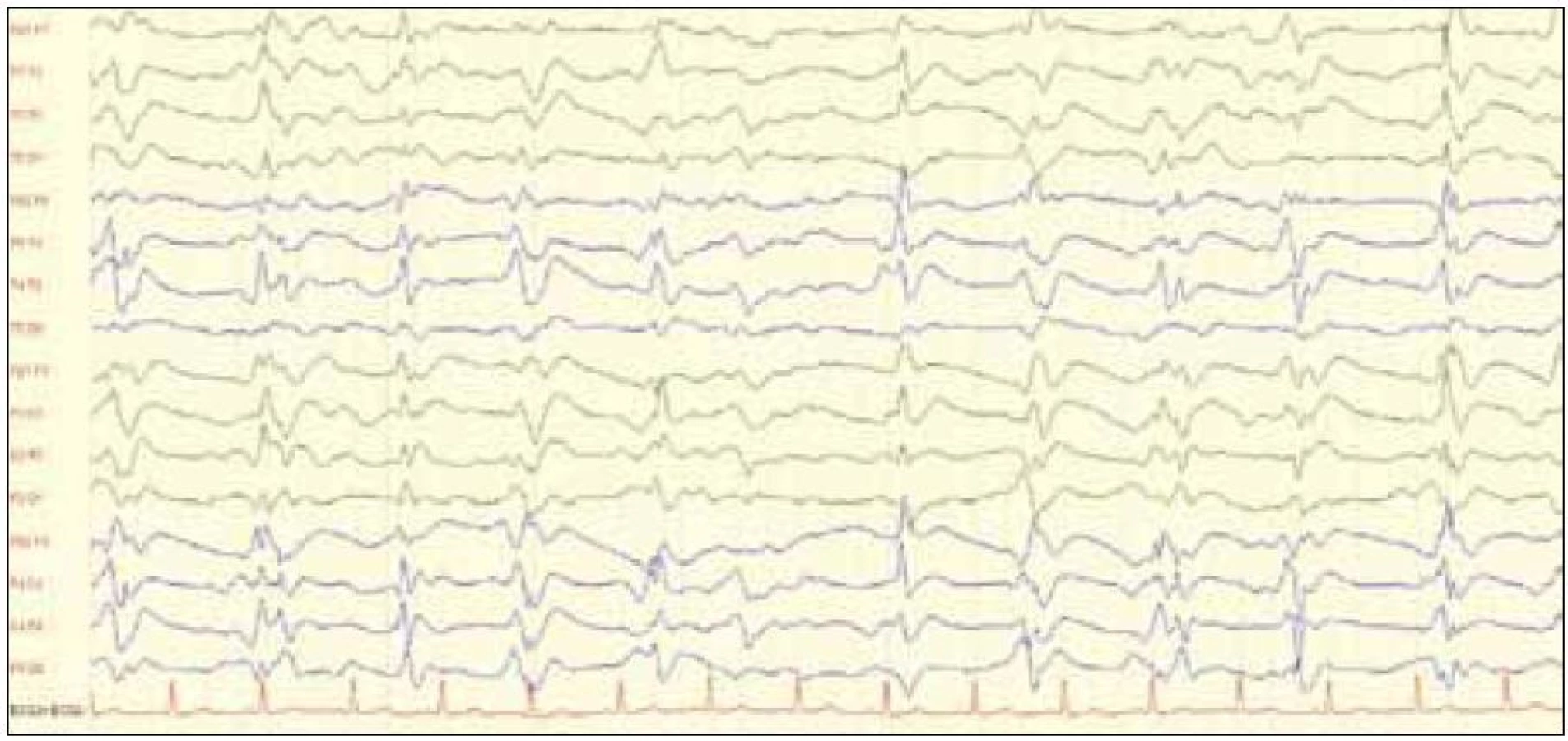
Ak sa trvanie ochorenia rozdelí na tretiny, prítomnosť typických PSWCs komplexov bola signifikantne nižšia v prvej tretine ochorenia ako v poslednej [33]. To však neplatí, ak sa zohľadní rozdelenie sCJch na jednotlivé subtypy. Typické EEG zmeny sú prítomné u subtypov s prevažne kortikálnym postihnutím, teda MM1 a MV1 [46].
Definitívnym potvrdením diagnózy je neuropatologické vyšetrenie.
Neuropatologické vyšetrenie
Definitívna diagnóza ľudských prionových ochorení je založená na neurohistologickom vyšetrení mozgového tkaniva doplnenom imunohistochemickými metódami, ktoré sa robia niekoľkými typmi protilátok a metódou westernblot, ktoré overujú prítomnosť patologicky zmeneného prionového proteinu v tkanive. V prípade familiárnych foriem sa sekvenuje gén PRNP a hľadá sa kauzálna patogénna variácia [18].
Podľa WHO sú podporným kritériom pre stanovenie diagnózy CJch (sporadickej, iatrogénnej alebo familiárnej formy) [24,25]:
- spongiformné zmeny v mozgovej a/alebo mozočkovej kôre a/alebo subkortikálnej sivej hmote a/alebo,
- encefalopatia s imunopozitivitou patologického PrP (difúzne synaptické zmeny a/alebo zmeny roztrúsené perivakuolárne a/alebo zmeny typu plakov.
Makroskopické zmeny
Makroskopické zmeny nie sú výrazné, najčastejšie býva atrofia, ktorá môže byť difúzna alebo viac lokalizovaná, hlavne v okcipitálnej, striatálnej a thalamickej oblasti a v cerebele [52]. Na rozdiel od Alzheimerovej choroby je oblasť hipokampu vcelku dobre zachovaná aj u pacientov so závažnou atrofiou.
Histopatológia
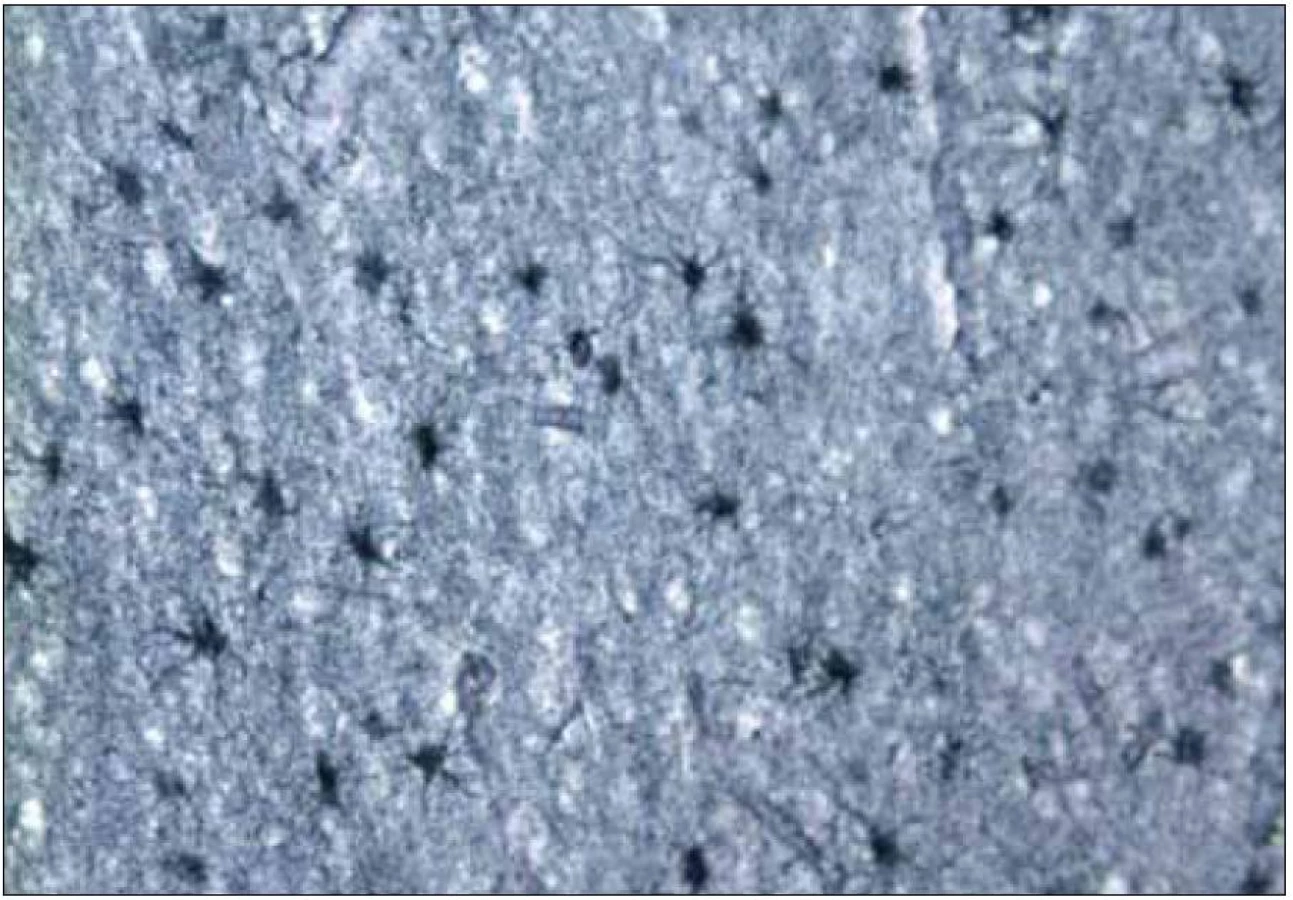
Pre diagnózu CJch sú z histopatologického hľadiska najtypickejšie spongiformné zmeny, numerická strata neurónov a glióza postihujúca tak astrocyty, ako aj mikrogliu (obr. 6, 7) [16,32]. Pre potvrdenie diagnózy je potrebné vyšetrenie vzoriek z viacerých oblastí mozgu (frontálneho, temporálneho a okcipitálneho laloka), bazálnych ganglií a cerebela. Dostatočné môže byť aj vyšetrenie jedného bločku tkaniva s typickými histologickými zmenami a/alebo jednoznačnou imunopozitivitou patologického PrP, ale len pre stanovenie CJch a nie podtypu ochorenia.
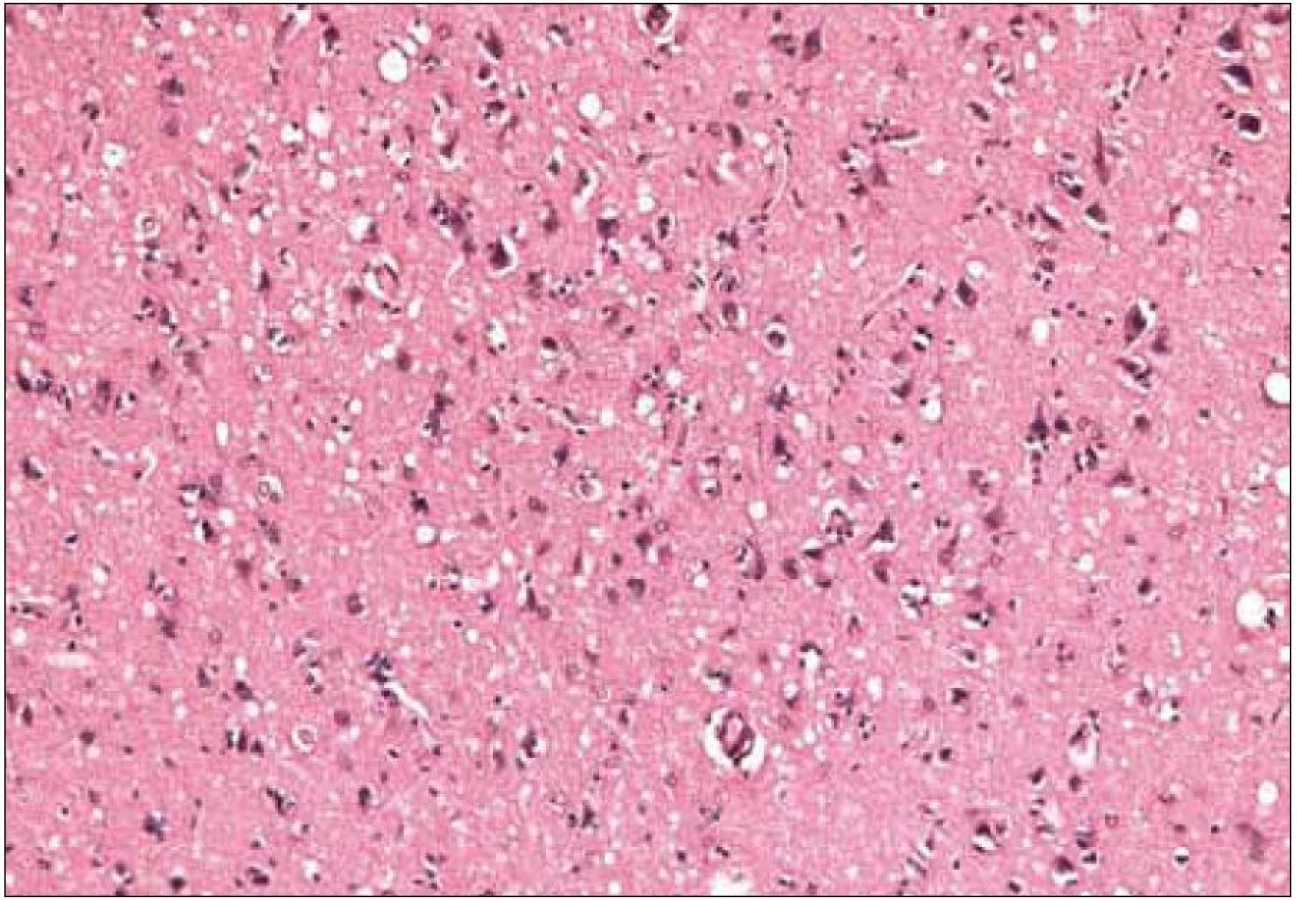
Kým strata neurónov alebo glióza sú časté aj u iných ochorení CNS, spongiformné zmeny sú relatívne špecifické pre CJch [32]. Bývajú prítomné v oblasti mozgovej kôry, v subkortikálnej sivej hmote a v mozočku. Vždy sú prítomné v hlave nucleus caudatus. Zriedkavo bývajú v oblasti mozgového kmeňa a v mieche, aj keď akumulácia patologického PrP môže byť aj v týchto oblastiach [32].
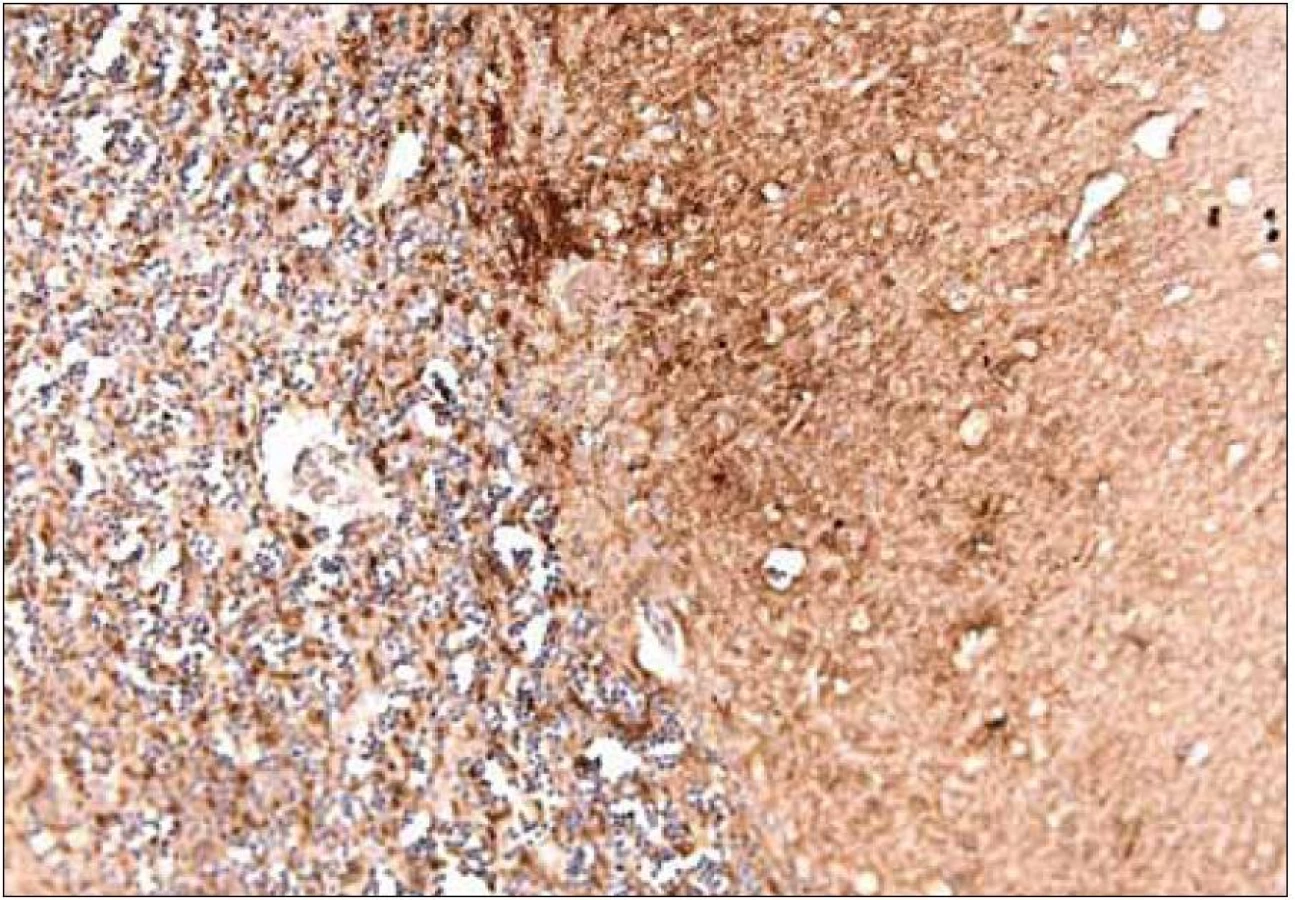
Rozhodujúcu úlohu pri stanovení diagnózy má imunohistochemické vyšetrenie, ktoré využíva rôzne typy protilátok na dôkaz patogénnych prionov v tkanive (obr. 8). Súčasne je biochemicky typizovaný subtyp prionového proteínu metódou Western blot [18,53,54].
Histopatologické a imunohistochemické vyšetrenie sa robí prevažne z materiálu získaného z autopsie. Biopsia mozgu sa robí zriedkavo, predovšetkým u pacientov, kde v diferenciálnej diagnostike prichádza do úvahy liečiteľná diagnóza. Otázka biopsie mozgu bude bližšie diskutovaná v časti diferenciálna diagnostika CJch, pretože úzko súvisí práve s touto problematikou.
Genetická forma Creutzfeldtovej-Jakobovej choroby
Ako genetická forma Creutzfeldtovej-Jakobovej choroby sa označuje prionové ochorenie podmienené mutáciou v géne pre PRNP, ktoré má klinické a histopatologické znaky spadajúce do spektra CJch. Ak sú v rodine viacerí postihnutí jedinci, hovoríme o familiárnej forme. Genetická forma tvorí 10–15 % ľudských prionových ochorení, ku krajinám s podstatne vyšším výskytom patrí Slovensko. Je známych najmenej 20 odlišných mutácií, ktoré sú všetky prenášané ako autozomálne dominantné a zahŕňajú bodové mutácie (napr. Asp178Asn s normálnym variantom Val129, mutácie Val180Ile, Thr183Ala, Glu196Lys, Glu200Lys, Val203Ile, Arg208His, Val210Ile, Glu211Gln, Met232Arg), delečné alebo inzerčné mutácie [55]. Delečné alebo inzerčné mutácie sú lokalizované medzi kodónom 51 a 91. Normálne PRNP alely sú tvorené jedným nonapeptidom a 4 opakujúcimi sa oktapeptidovými sekvenciami, pričom každá pozostáva z nasledovných aminokyselín: Pro-(His/Gln)-Gly-Gly-Gly-(-/Trp)-Gly-Gln. U pacientov s CJch bola identifikovaná inzercia ďalších jeden až deväť oktapeptidov. Inzercia štyroch oktapeptidov nebola doposiaľ asociovaná s prionovým ochorením. Sporadický výskyt jednej oktapeptidovej delécie je považovaný za normálny polymorfizmus, delécia 2 oktapeptidov je patologická [16,55].
Genetická forma CJch zodpovedá klinickým obrazom, histopatologickými zmenami, začiatkom a trvaním ochorenia a EEG zmenami jednotlivým fenotypom sporadickej CJch, i keď pre niektoré mutácie je typický skorší začiatok a dlhšie trvanie ochorenia a existujú aj určité rozdiely v histopalogickom obraze. U genetických foriem s dlhšími inzerčnými mutáciami sú spongiformné zmeny minimálne alebo chýbajú a non-amyloidové plaky tvorené patologickým PrP sú zriedkavé, kuru plaky boli popísané doteraz len u dvoch foriem genetickej CJch [16,55].
Najčastejšie haplotypy
Najčastejšou mutáciou je E200K-129M haplotyp. Najväčší súbor bol zaregistrovaný u líbijských a tuniských židov [56], unikátne geografické zoskupenie sa vyskytuje na Slovensku (hlavne na Orave, Kysuciach a v okolí Rožňavy), kde sa vôbec prvý raz potvrdil špecifický vzťah mutácie E200K ku vzniku CJch [5,57] a v Chile. Boli zaregistrované aj individuálne rodiny alebo prípady bez pozitívnej rodinnej anamnézy [55–57].
Klinický obraz
Klinický obraz haplotypu E200K-129M je porovnateľný so sporadickou formou, s najčastejším subtypom MM1, teda najčastejšie je prítomný kognitívny deficit, psychické zmeny, ďalej cerebelárne príznaky, zrakové problémy a myoklonické zášklby a nakoniec rozvoj demencie. Typické periodické komplexy ostrých vĺn na EEG sú prítomné asi u 74–76 % pacientov v porovnaní s 80 % u sporadickej formy, aj prítomnosť proteínu 14-3-3 a celkového tau proteínu v likvore má o niečo nižšiu senzitivitu ako u sporadickej formy [42]. Jediným typickým príznakom, ktorý by mohol odlíšiť túto formu od iných foriem, je postihnutie periférneho nervového systému, ktoré je len zriedkavo pozorované u sCJch MM1. Polyneuropatia je axonálne demyelinizačná so segmentálnou demyelinizáciou a často je sprevádzaná aj zvýšením hladiny bielkovín v likvore [58,59]. Histologické zmeny sú taktiež podobné ako u sCJch MM1.
E200K-129V haplotyp
Prvá rodina bola popísaná v Rakúsku [60], klinický obraz a histopatologické zmeny pripomínajú najviac subtyp sCJch VV2, teda v úvode je prítomná ataxia, myoklonus a EEG zmeny sa objavujú až v neskorších štádiách ochorenia.
D178-129V haplotyp
U haplotypu D178N-129V sa jedná o mutáciu PRNP v kodóne 178 (D178N), ktorá v prípade, že sa nachádza na alele, ktorá má v kodóne 129 valín, spôsobuje familiárnu CJch. Rodiny s týmto haplotypom boli popísané v USA (s pôvodom aj v iných krajinách ako USA), Francúzsku, Fínsku a Nemecku, 3 rodiny sú známe aj v ČR. Začiatok ochorenia je skorší, okolo 39 rokov, a trvanie kratšie, ak je prítomný homozygotný genotyp VV v kodóne 129 oproti heterozygotom, kedy je začiatok okolo veku 49 rokov [61,62] a trvanie ochorenia dlhšie, okolo 27 mesiacov. V klinickom obraze dominujú poruchy pamäti, správania, neskôr sa pridružuje ataxia, poruchy reči, tremor a myoklonus, môžu byť prítomné zmeny na EEG [63].
Pre dlhšie trvanie ochorenia a príznaky ktoré môžu imitovať niektoré atypické demencie, najmä frontotemporálnu demenciu, je potrebné ich odlíšiť v diferenciálnej diagnostike.
V prípade, že sa mutácia PRNP v kodóne 178 (D178N) nachádza na alele, ktorá má v kodóne 129 metionín, vzniká familiárna fatálna insomnia.
Z genetických foriem prionových ochorení bol v ČR popísaný výskyt Gerstmannovho-Sträusslerovho-Scheinkerovho syndrómu, mutácie E200K, D178N a vzácnej mutácie R208H [62,64].
Nový variant Creutzfeldtovej-Jakobovej choroby
Nový variant Creutzfeldtovej-Jakobovej choroby (vCJch) bol prvýkrát popísaný vo Veľkej Británii v roku 1996 [65,66], kedy ochoreli mladí ľudia s priemerným vekom začiatku ochorenia 28 rokov a odlišným priebehom a dlhším trvaním ochorenia ako u dovtedy známej CJch. Do roku 2011 bolo popísaných 174 prípadov, z toho najviac vo Veľkej Británii, ale aj vo Francúzsku, Španielsku, Írsku, Nizozemsku, USA, Portugalsku, Taliansku, v Kanade, na Taiwane, v Saudskej Arábii a v Japonsku. vCJch súvisí s epidémiou bovinnej spongiformnej encefalopatie (BSE) a k ochoreniu najpravdepodobnejšie došlo nákazou prionmi chorých zvierat konzumáciou kontaminovaného mäsa. Vzťah vCJch k BSE určila typizácia kmeňa prionov a experimentálny prenos na transgénnu aj konvenčnú myš [18]. Neskôr bol zaznamenaný prenos vCJch krvnou cestou – transfúziou krvných produktov [67,68].
Zásadným rozdielom oproti klasickému variantu je aj fakt, že pôvodca nákazy bol zistený aj v periférnych lymfatických orgánoch, čo významne zvyšuje riziko prenosu. Všetci doteraz diagnostikovaní pacienti s vCJch boli v kodóne 129 homozygoti pre metionín [69]. U žiadneho pacienta s vCJch nebola zistená mutácia génu pre PRNP. Za rizikové faktory vCJch sa preto považujú: konzumácia hovädzieho mäsa, homozygozita pre metionín v kodóne 129 génu priónového proteínu, vek mladší ako 55 rokov, krvná transúzia a pobyt vo Veľkej Británii [65].
Diagnostika
Kritériá pre diagnózu nového variantu CJch sú uvedené v tab. 4 [70].
![Diagnostické kritériá pre nový variant Creutzfeldtovej-Jakobovej choroby (vCJch) – definitívna, pravdepodobná, možná [70].](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image/78b075d3be073421e2d355ab5fcafe99.png)
Patologickú formu PrP možno na rozdiel od sCJch dokázať v biopsii orgánov retikuloendotelového systému, hlavne v tkanive krčnej mandle a apendixu, preto v diagnostike vCJch je prínosom biopsia tonzíl, hlavne u pacientov, kde pri podozrení na vCJch je negatívny MR nález mozgu.
V diferenciálnej diagnostike prichádzajú do úvahy Alzheimerova choroba, cerebrovaskulárne ochorenia, vaskulitída, vírusová encefalitída a limbická encefalitída.
Klinický obraz
U väčšiny pacientov začína ochorenie psychiatrickými príznakmi, najčastejšie depresiou a úzkosťou [71]. Neurologické príznaky sa vyvíjajú asi po 6 mesiacoch od začiatku psychiatrických príznakov, u niektorých pacientov sa už v skoršej fáze ochorenia objavujú senzitívne príznaky – bolestivé parestézie a dysestézie, bolesti končatín bývajú niekedy bez iných senzitívnych porúch. Postupne pribúda dysartria, poruchy rovnováhy a zabúdanie. Dominantným príznakom u všetkých pacientov s vCJch je ataxia, v neskoršom priebehu aj s pádmi, a u pacientov s plne rozvinutým klinickým obrazom sa objavujú mimovoľné pohyby – myoklonus, chorea a dystónia. V terminálnom štádiu dominuje progresívne zhoršovanie kognitívneho deficitu, ktoré vyústi do demencie a často je prítomný akinetický mutizmus. Dľžka trvania ochorenia je v priemere 14 mesiacov (6–39 mesiacov).
Zobrazovacie metódy
Pre vCJch je na MR typický obraz abnormality v zadnej časti thalamu, ktorá sa najlepšie ukáže vo FLAIR a DWI zobrazení, ale možno ju pozorovať aj v T2 váženom MR obraze (obr. 9) a označuje sa ako príznak pulvinar thalami [72]. Má vysokú senzitivitu a špecificitu v koreláte s príslušným klinickým obrazom, a preto je tento príznak aj súčasťou aktuálnych kritérií pre vCJch [70]. U niektorých pacientov sa oblasť hyperintenzity šíri aj do oblasti dorzomediálneho jadra thalamu a do okolia aqueduktu na úrovni mozgového kmeňa.
![FLAIR MR s hyperintezitou v oblasti pulvinar thalami u pacienta s novým variantom CJch (vCJch) [72].
Publikované so súhlasom Elsevier Limited. Nakoľko sa vCJch v SR nevyskytol, obrázok je z literatúry.](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image/3a533dc58ddcd404bf5f74e642b596d8.jpg)
Laboratórne nálezy
U väčšiny pacientov sa na EEG zobrazuje nešpecifická pomalovlnová aktivita, ale EEG môže byť u vCJch aj negatívne. Yamada et al [73] a Binelli et al [74] popísali u dvoch pacientov v terminálnom štádiu typické periodické trifázické komplexy na EEG, typické pre sCJch. Základné vyšetrenie mozgomiechového moku môže byť negatívne, alebo môžu byť zvýšené proteíny. Proteín 14-3-3 je prítomný len asi u 1/2 pacientov s vCJch na rozdiel od sCJch, kde je to až u 80 % pacientov. Preto u pacientov s vCJch toto vyšetrenie nie je natoľko nápomocné v úvodných štádiách ochorenia [49], na rozdiel od prítomnosti tau proteínu v likvore.
U väčšiny pacientov nebývajú postmortem prítomné výrazné makroskopické zmeny na hemisférach mozgu, v oblasti mozgového kmeňa a mozočka, u pacientov s dlhším trvaním ochorenia býva prítomná atrofia cerebela, hlavne v oblasti vermis, a mierna atrofia mozgovej kôry, hlavne v okcipitálnej oblasti.
Neuropatologický nález pri vCJch sa od zmien pri sCJch líši histologicky, imunohistochemicky a imunologicky. Novú variantu charakterizuje veľký počet tzv. floridných plakov (početné amyloidové plaky s eozinofilnou centrálnou časťou a svetlou perifériou s lemom) v kôre mozgu a mozočka, spongiformné zmeny, strata neurónov, astrocytóza [65,66]. Imunohistochemickou metódou bol patologický PrP dokázaný hlavne v kôre mozgových hemisfér a mozočka, ale aj v ďalších častiach CNS, pozitívne farbenie na patologický PrP však bolo prítomné aj v niektorých periférnych nervových štruktúrach [75], ďalej v sietnici, zrakovom nerve, gangliách zadných koreňov a gangliách n. V, ale aj v lymfatických uzlinách, uzlinách v oblasti brucha, slezine a týme.
Iatrogenná Creutzfeldtova-Jakobova choroba
Iatrogénna Creutfeldtova-Jakobova choroba (iCJch) sa v súčasnosti označuje ako náhodne prenesená (accidentally transmitted) a vzniká prenosom prionového ochorenia z človeka na človeka.
Prvý prípad iCJch bol popísaný v roku 1974 u príjemcu transplantovanej rohovky od zomrelého darcu, u ktorého bola potvrdená sCJch [17]. Do apríla 2010 bolo registrovaných celkovo 420 prípadov iCJch, Brown et al [76] v júni 2012 publikovali novšie údaje, prenos kontaminovaným rastovým hormónom u 226 a štepmi dura mater z kadaverov s nediagnostikovanou CJch u 228 pacientov. Najviac prípadov iCJch po použití štepov dura mater bolo pri prípravku Lyodura od nemeckého výrobcu vyrobeného pred rokom 1987 [76]. Niekoľko ďalších prípadov bolo zapríčinených kontamináciou neurochirurgickými nástrojmi, elektroencefalografickými elektródami, rohovkovými štepmi a gonadotropným hormónom.
Dodržiavaním preventívnych opatrení pri chirurgických zákrokoch, označením biologicky nebezpečných vzoriek, správnym opatrením pri čistení a dekontaminácii nástrojov by mal byť iatrogénny prenos obmedzený na najnižšiu možnú mieru. Na Slovensku ani v ČR zatiaľ nebol registrovaný prípad vCJch. V ČR sa od 1. 1. 2007 povinne testuje mozgové tkanivo všetkých darcov rohoviek na prítomnosť patologickej formy PrP metódou Western blot, vyšetrených bolo 1 142 darcov s negatívnym výsledkom [77]. Na Slovensku bolo testovaných 1 133 darcov rohoviek a 970 kontrol na prítomnosť mutácie E200K v géne pre PRNP, mutácia E200K bola zistená u dvoh donorov a štyroch jedincov kontrolného súboru, 48 % zo všetkých vyšetrených boli v kodóne 129 homozygoti pre metionín [78].
Inkubačná doba iCJch je všeobecne dlhá, údaje sa pohybujú od 1,3 do 42 rokov, ale nikdy nebola kratšia ako 12 mesiacov. Inkubačná doba je u intracerebrálnej nákazy kratšia (mesiace) ako u periférnej nákazy (roky). Rizikovým faktorom pre vznik je homozygozita MM alebo VV, homozygoti majú aj kratšiu inkubačnú dobu [5,17,76].
Diagnostika
Diagnostické kritéria pre iatrogénnu CJch sú v tab. 5 [17,25]. V diferenciálnej diagnostike je potrebné myslieť na všetky ochorenia s progresívnou cerebelárnou symptomatológiou a ochorenia s rýchle progredujúcim kognitívnym deficitom.
![Diagnostické kritériá pre iatrogénnu Creutzfeldtovu-Jakobovu chorobu – definitívna, pravdepodobná [17,25].](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image/96838ab1e6ce2d72465714fe5286527f.png)
Klinický obraz
Klinický obraz pripomína sCJch alebo vCJch, dominuje progresívna ataxia, ďalšie príznaky vrátane demencie sa objavujú neskôr alebo môžu chýbať [79,80].
Pomocné vyšetrenia na diagnostiku iCJch, neuropatologické a mikroskopické nálezy, ako aj nálezy pri MR mozgu sú podobné ako pri sCJch, v likvore môžu byť nešpecificky zvýšené bielkoviny, neboli robené štúdie sledujúce prítomnosť proteínu 14-3-3 v likvore u pacientov s iCJch, ale pozitívny nález bol u pacientov po liečbe rastovým hormónom.
Diferenciálna diagnostika Creutzfeldtovej-Jakobovej choroby
V rámci diferenciálnej diagnostiky CJch zvažujeme všetky rýchlo progredujúce demencie (RPD). Zo skupiny neurodegeneratívnych ochorení sú to predovšetkým Alzheimerova choroba (Ach), frontotemporálna demencia (FTD), kortikobazálna degenerácia (KBD) a demencia s Lewyho telieskami (DLB), hlavne pri ich fulminantnom priebehu [81]. Významnú skupinu tvoria autoimúnne podmienené demencie, ktoré sú liečiteľné [82]. Je nad rámec tejto práce podrobne rozvíjať diferenciálnu diagnostiku autoimúnne podmienených demencií, z menej známych treba však spomenúť 1. Autoimúnnu limbickú encefalitídu (LE), ktorá môže byť paraneoplastická (PLE) alebo neparaneoplastická (NLE). V klinickom obraze väčšinou dominuje mnestický deficit, parciálne alebo generalizované epileptické záchvaty, psychiatrické príznaky (bludy, halucinácie, depresia, zmeny osobnosti a chovania) [83–86]. Diagnostika sa opiera aj o MR mozgu, EEG a likvorový nález, typická býva výrazná hyponatrémia. NLE je spájaná s protilátkami proti povrchovým bunečným antigénom, konkrétne napäťovo-riadeným káliovym kanálom (VGKC), NMDA receptorom (NMDAR), AMPA receptorom (AMPAR) a ďalším [87]. Z hľadiska prognózy pacienta je pre ňu typický dobrý terapeutický efekt vysokých dávok kortikoidov. Niektorí autori popisujú aj efekt plazmaferézy alebo intravenóznych imunoglobulínov [81,86–88]. Treba upozorniť, že asi u 20 % NLE s protilátkami proti VGKC sa môže dokázať prítomnosť nádorového ochorenia. PLE je najčastejšie spojená s malobunkovým karcinómom pľúc, karcinómom prsníka a semenníkov, tymómom, lymfómom. Diagnostika paraneoplastickej limbickej encefalitídy sa opiera o dôkaz paraneoplastických protilátok proti intracelulárnym antigénom (anti-HU, anti-MA2, anti-CV) [81,87]. 2. Encefalopatiu reagujúcu na liečbu steroidmi asociovanú s autoimúnnou tyreoiditídou – Steroid-Responsive Encephalopathy Associated with autoimmune Thyroiditis (SREAT), ktorá sa niekedy označuje aj Hashimotova encefalopatia. V čase objavenia sa neurologických príznakov nemusia byť prítomné klinické ani biochemické prejavy ochorenia štítnej žľazy, na potvrdenie diagnózy majú byť prítomné antityreoidálne protilátky: anti-tyreoglobulín (anti-Tg) alebo sérová anti-tyreoperoxidáza (anti-TPO). V klinickom obraze dominujú fluktuujúce kognitívne poruchy, tremor, myoklonus a neuropsychiatrické príznaky. Niekedy môže byť prítomný EEG nález typický pre CJch, MR nezobrazuje zmeny typické pre CJch, čo pomáha v diferenciálnej diagnostike [39,40,89]. Vysoké dávky intravenóznych kortikosteroidov (metylprednizolonu) vedú u niektorých pacientov k efektu v priebehu niekoľkých týždňov, u niektorých v priebehu niekoľkých dní [83]. Z ďalších liečiteľných RPD sú vaskulitídy CNS, ktoré sa od CJch líšia hlavne MR nálezom, s početnými infarktmi mozgu alebo mozgovým krvácaním [90]. V diferenciálnej diagnostike sú problémom predovšetkým primárne vaskulitídy CNS, ktoré sú ale zriedkavým ochorením CNS. Klinické príznaky sú nešpecifické – bolesti hlavy, encefalopatia, dľžka trvania ochorenia je rozdielna. Pomocné vyšetrenia (serológia, likvor, angiografia, zobrazovacie vyšetrenia) môžu byť negatívne, a preto pre stanovenie definitívnej diagnózy je často potrebná biopsia mozgu. Reagujú na liečbu imunosupresívami [81,90,91].
Ataxiu, neuropsychiatrické príznaky, neuropatiu a demenciu, aj pri absencii gastrointestinálnych príznakov, môže niekedy spôsobovať aj celiakia, diagnóza sa opiera o zvýšenú hladinu antiendomysiálnych alebo antigliadínových imunoglobulínov A (IgA) alebo IgG. Diagnóza sa potvrdzuje biopsiou tenkého čreva s imunohistochemickým vyšetrením. Liečbou je bezgluténová diéta [92].
V diferenciálnej diagnostike treba myslieť aj na sarkoidózu a vaskulárnu demenciu [81].
Ďalšiu veľkú skupinu ochorení v diferenciálnej diagnostike predstavujú vírusové a bakeriálne infekcie CNS – vírusové encefalitídy, neurosyfilis, neuroborelióza, kde sa diferenciálna diagnostika opiera predovšetkým o likvorový nález [93–96]. U ľudí „cestovateľov“ treba myslieť aj na parazitárne ochorenia, predovšetkým tryponosomiázu a maláriu [97].
Klinický obraz RPD môže byť spôsobený aj nádorovým ochorením. Väčšina nádorov sa dá ľahko diagnostikovať MR vyšetrením mozgu, problémom môžu byť primárny lymfóm CNS a gliomatóza mozgu, a to z hľadiska stanovenia diagnózy týchto ochorení, ktorá častokrát vyžaduje biopsiu mozgu [98–100].
Biopsia mozgu predstavuje nielen medicínsky, ale aj etický problém. Vždy ju treba zvažovať s prihliadnutím na prínos jej výsledku pre ďalšiu prognózu pacienta a na riziká spojené s týmto vyšetrením. So zdokonaľovaním zobrazovacích vyšetrení a laboratórnej diagnostiky, predovšetkým dôkazu protilátok u liečiteľných autoimúnne podmienených rýchle progredujúcich demencií, sa počet biopsií mozgu výrazne znížil. Biopsia mozgu u pacientov s demenciou sa dnes robí zriedkavo, predovšetkým u pacientov, kde v diferenciálnej diagnostike prichádza do úvahy liečiteľná diagnóza. Schott et al [101] publikovali analýzu počtu biopsií a najčastejších príčin biopsie u pacientov s demenciou v roku 2005 (z bioptického materiálu za roky 1989–2003) a v roku 2010 (z bioptického materiálu za roky 2004–2009) a skonštatovali výrazný pokles ročného počtu biopsií, a to zo 6/rok na 3/rok. Zlepšila sa aj zhoda suponovanej diagnózy s výsledkom biopsie. V prvom sledovanom období bola najčastejším dôvodom biopsie mozgu Alzheimerova choroba, potom CJch a následne zápalové ochorenia, v druhom sledovanom období bola najčastejšou príčinou CJch. Všetky zistenia dávajú do súvisu práve so zlepšenou diagnostikou demencií. Pre úspešný výsledok biopsie mozgu je potrebný odber dostatočne veľkej vzorky, ktorá má obsahovať celú hrúbku mozgovej kôry so subkortikálnou bielou hmotou a priľahlými obalmi mozgu, súčasne je však potrebné prihliadať k tomu, aby odber vzorky nespôsobil zhoršenie neurologického deficitu pacienta [101]. Pri biopsii mozgu u pacienta s podozrením na CJch musia byť dodržiavané nariadenia manipulácie s infekčným materiálom, doporučený pracovný postup pre ošetrovanie pacientov s podozrením na CJch, vCJch a režim dekontaminácie a sterilizácie [102] a pokyn „Pitvy a bioptické vyšetrenia mozgu pri podozrení na prionové choroby“ [103,104].
Zásady pre bioptické vyšetrenia mozgu, chirurgické výkony a dekontamináciu a sterilizáciu pri podozrení na prionové choroby
Ak sa jedná o mozgovú biopsiu pri nejasnom ochorení, keď prichádza do úvahy aj možnosť prionovej choroby, operujúci lekár v spolupráci s miestnym patológom rozdelí odobrané tkanivo na dve časti. Jedna časť sa fixuje štandardne a vyšetrí ju miestny patológ. Druhá časť je nefixovaná a zmrazená na suchom ľade sa dopraví do Národného referenčného laboratória (NRL) (v ČR alebo SR), kde sa urobí vyšetrenie metódou Western blot. Táto metóda vyžaduje len malý objem tkaniva, je citlivejšia než obvyklá imunohistochémia a určí prítomnosť alebo neprítomnsť prionov v tkanive. Tento výsledok sa oznámi miestnemu patológovi. Ak je výsledok negatívny, zaháji miestny patológ vyšetrovanie biopsie. Ak je výsledok pozitívny, miestny patológ obratom pošle materiál do NRL za účelom následného neurohistologického a imunohistochemického vyšetrenia [103].
Biopsia aj operačné výkony u pacientov s podozrením na CJch podliehajú špeciálnym predpisom, ktorých podrobný popis je nad rámec tejto práce. K základným zásadám patrí plánovanie operačných výkonov na koniec operačného programu, používanie jednorázových nástrojov a pomôcok, pokiaľ je to možné, ak sa používajú nástroje a pomôcky na opakované použitie, používajú sa len termostabilné, autoklávovateľné a odolné voči použitiu 4% NaOH alebo 5,25% chlornanu sodného. Odobraný materiál sa označí ako „biologicky nebezpečný“.
Priony sú neobvykle rezistentné voči dezinfekcii a sterilizácii bežnými fyzikálnymi a chemickými metódami, ktoré sa používajú pre dekontamináciu infekčných patogénov, preto pri podozrení na prionové ochorenie musia byť dodržané určené zásady dekontaminácie a sterilizácie. Najbezpečnejšou metódou prevencie rizika reziduálnej infekcie na kontaminovaných nástrojoch a materiáloch je likvidácia a zničenie spálením. Odpad sa umiestňuje do plastových vriec s označením „biologicky nebezpečené“, ktoré musia byť neodkladne spálené. Nástroje a ostatné predmety a materiály určené k opakovanému použitiu musia byť udržiavané vlhké od okamihu ich kontaktu s infekčným materiálom až do následného očistenia a dekontaminácie. Osoby, ktoré robia dekontamináciu, musia mať jednorázové ochranné prostriedky [102].
Usmernenia pre ošetrovanie pacientov s podozrením na CJch
Každý aj suspektný prípad prenosnej spongiformnej encefalopatie podlieha povinnému hláseniu. Bežný sociálny a klinický kontakt a neinvazívne klinické vyšetrenia u pacientov s podozrením na CJch nepredstavujú pre zdravotníckych pracovníkov, príbuzných alebo spoločnosť riziko. Nie je potrebná izolácia pacientov. Nie sú potrebné žiadne špeciálne opatrenia pri manipulácii s príbormi, s hadičkami určenými na kŕmenie alebo s prádlom. Pacienti môžu absolvovať vyšetrenia bežnými diagnostickými postupmi. V maximálnej miere sa majú používať jednorázové pomôcky, ak to nie je možné, pomôcky sa majú individualizovať pre každého pacienta s podozrením na CJch. Zvláštnu pozornosť je potrebné venovať lumbálnej punkcii, vyšetrujúci používa jednorázové ochranné pomôcky, používa sa materiál a pomôcky na jedno použitie, použitý materiál sa zhromaždí v kontajneroch s označením „biologicky nebezpečný“ a zabezpečí sa jeho urýchlená likvidácia spálením [102–104].
Liečba
Všetky formy Creutzfeldtovej-Jakobovej choroby sú fatálne, preto neexistujú v súčasnosti žiadne terapeutické postupy pre liečbu CJch. Doteraz bolo testovaných viacero liekov ako amantadin, steroidy, interferóny, acyklovir, protivírusové lieky, antibiotiká a aj v súčasnosti prebieha testovanie ďalších liekov [105].
Mierny efekt na priebeh ochorenia bol zaznamenaný u quinacrinu, flupirtinu a doxycyclinu. V pokusoch na tkanivových kultúrach sa ukázalo, že Quinacrine má potenciál zabrániť formovaniu PrPSC a dokonca ho odstrániť z postihnutých buniek. Jeho výhodou je, že prechádza hematoencefalickou bariérou. Kontrolované štúdie však zatiaľ nepreukázali jeho vplyv na dľžku ochorenia [106].
Pozitívnejšie výsledky boli pozorované s flupirtinom (amidopyridin), centrálne pôsobiacim nonopiátovým analgetikom, ktorý síce takisto neovplyvnil dľžku prežívania, ale mierne zlepšil výkon v kognitívnych testoch [107].
Najlepšie výsledky boli dosiahnuté s doxycyclinom, ktorý, na základe experimentálnych výsledkov, sa viaže priamo na PrPSC, čím zabraňuje ďalším konformačným zmenám. V experimente predľžil čas prežívania škrečkov. Následne boli realizované štúdie u pacientov v Taliansku a Nemecku s dávkou 100 mg denne, ktoré potvrdili spomalenie progresie ochorenia. V súčasnosti sa doxycyclin skúša ako profylaktická liečba u zdravých nosičov mutácie [105,108].
U väčšiny pacientov sa používa symptomatická liečba zameraná predovšetkým na liečbu bolesti a porúch správania. Cieľom liečby je zlepšiť ich kvalitu života. Pacienti často trpia aj úzkosťou, depresiami, niekedy psychotickými príznakmi, v liečbe ktorých sa uplatňujú hlavne anxiolytiká a antipsychotiká. Keďže pacienti majú často aj extrapyramídové príznaky, uprednostňujeme atypické antipsychotiká pre menší vplyv na zhoršenie extrapyramídových príznakov. Na zmiernenie myoklonu sa odporúča klonazepam alebo valproát sodný v štandardnej dávke [105].
V terminálnom štádiu ochorenia uprednostňujeme ošetrovateľskú a paliatívnu starostlivosť v spolupráci s rodinou pacienta [109].
Etické problémy a paliatívna starostlivosť u pacientov s Creutzfeldtovou-Jakobovou chorobou
K závažným etickým problémom u familiárnych foriem CJch patrí genetické testovanie rodinných príslušníkov. Každý príbuzný si vyžaduje individuálny prístup, je potrebné mu vysvetliť, že môže byť nosičom mutácie a opýtať sa ho, či chce byť testovaný. Takisto je potrebné mu vysvetliť, že pri pozitívnom výsledku testovania je veľmi vysoká pravdepodobnosť že ochorie, pretože penetrácia génu je vysoká [110]. Z našej skúsenosti môžem povedať, že sa väčšina príbuzných pacientov s potvrdenou genetickou formou ochorenia odmietla testovať. Toto ich rozhodnutie je potrebné rešpektovať. Genetické testovanie predstavuje závažný zásah do života vyšetrovaného, predovšetkým poznanie pozitívneho výsledku má zásadný vplyv na jeho ďalší život. Preto si vyžaduje spoluprácu neurológa, klinického genetika, psychiatra a psychológa. Po úvodnom vysvetlení problematiky a následkov pozitívneho testovania by mal pacient dostať určitý čas, aby sa mohol rozhodnúť, či chce testovanie podstúpiť. Musí to byť jeho rozhodnutie, pretože on bude znášať všetky dôsledky tohto rozhodnutia.
Ďalším etickým aspektom ochorenia je starostlivosť o pacienta s CJch v terminálnom štádiu ochorenia. Vzhľadom na prognózu ochorenia je v tomto štádiu dôležité zabezpečiť pacientovi dôstojné dožitie, nezaťažovať ho náročnými diagnostickými výkonmi a terapeutickými a ošetrovateľskými postupmi (napríklad perkutánna gastrostómia – PEG), ktoré nezvrátia priebeh ochorenia, ale môžu zhoršiť utrpenie pacienta. Každý pacient by mal spolurozhodovať o svojej liečbe a starostlivosti, avšak pacient v terminálnom štádiu CJch toho nie je schopný, preto je najlepšie vždy zvoliť tímový spôsob rozhodovania aj v spolupráci s príbuznými pacienta. Postup v terminálnom štádiu je podobný ako paliatívna starostlivosť o pacienta s demenciou, ktorý publikovali Rusina et al [109].
Záver
Creutzfeldtova-Jakobova choroba je v súčasnosti smrteľné ochorenie, u ktorého definitívne potvrdenie diagnózy je možné len neuropatologickým vyšetrením post mortem alebo za života biopsiou mozgu. U pacientov s podozrením na CJch sa robí ešte počas života genetické testovanie, ktoré je nápomocné pri stanovení diagnózy genetickej formy CJch. Jeho význam v SR a ČR je o to väčší, že je tu zmapovaný nadmerný výskyt genetickej formy s mutáciou E200K v SR a v ČR a okolitých krajinách bol zaznamenaný výskyt Gerstmannovho-Sträusslerovho-Scheinkerovho syndrómu – familárnej formy prionového ochorenia.
Klinický obraz a priebeh ochorenia môže byť rôzny, od obrazu typického pre sporadickú formu až po rôzne atypické formy. Vzhľadom k tomu, že podobný klinický obraz môžu mať niektoré liečiteľné, rýchle progredujúce demencie, je potrebné na nich v diferenciálnej diagnostike myslieť, diagnostikovať ich a včas zahájiť liečbu.
Správne rozpoznanie prionového ochorenia má zásadný význam pre pacienta s týmto ochorením a jeho rodinu. Keďže neexistuje kauzálna liečba, pacienta netreba zaťažovať náročnými pomocnými vyšetreniami, ale zahájiť včasnú paliatívnu liečbu.
Autor deklaruje, že v souvislosti s předmětem studie nemá žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
prof. MUDr. Zuzana Gdovinová, CSc.
Neurologická klinika LF UPJŠ a UN LP Košice
Trieda SNP 1
040 01 Košice
e-mail: zuzana.gdovinova@upjs.sk
Prijato k recenzii:17. 12. 2012
Prijato do tlače: 11. 3. 2013
Recenzenti
doc. MUDr. Radoslav Matěj, Ph.D.
MUDr. Eva Mitrová, DrSc.
MUDr. Robert Rusina, Ph.D.
Ďakujem MUDr. Eve Mitrovej, DrSc., z Národného referenčného centra pre prionové choroby a Ústavu preventívnej a klinickej medicíny SZU, Bratislava, MUDr. Eve Feketeovej, PhD., z Neurologickej kliniky LF UPJŠ a UN LP Košice, RNDr. Viere Habalovej, PhD., z Ústavu biológie LF UPJŠ, MUDr. D. Kozákovej, MUDr. M. Andrašinovej a MUDr. Z. Lehotskej z Kliniky radiologie a zobrazovacích metód UN LP Košice za spoluprácu a poskytnutie obrazovej dokumentácie.
prof. MUDr. Zuzana Gdovinová, CSc.

Zuzana Gdovinová ukončila štúdium na Lekárskej fakulte Univerzity P. J. Šafárika v Košiciach v roku 1984. Odvtedy pracuje na Neurologickej klinike LF UPJŠ a UN LP Košice, od roku 2003 ako prednostka kliniky. V roku 1995 obhájila na LF UK Bratislava kandidátsku prácu: „Význam transkraniálneho ultrazvukového vyšetrenia mozgových ciev dopplerovou metódou pre klinickú prax“, v roku 2003 bola po obhájení habilitačnej práce „Prínos transkraniálneho ultrazvukového vyšetrenia mozgových ciev (TCD) pri posudzovaní alkoholu ako rizikového faktora ischemických cievnych mozgových príhod“, takisto na LF UK Bratislava, menovaná docentkou neurológie a v roku 2012 bola na návrh Vedeckej rady UK v Prahe menovaná profesorkou pre odbor neurológia. V oblasti klinickej práce a výskumu sa venuje problematike cievnych ochorení mozgu a problematike demencií, je zástupcom Slovenska v panely EFNS „Dementia and cognitive neurology“. Klinika je súčasťou Centra excelentnosti pre výskum aterosklerózy. Podieľa sa na riešení viacerých grantových úloh. Dlhodobo spolupracuje pri výchove doktorandov s univerzitou v Groningene (Nizozemsko). Je autorkou alebo spoluautorkou 1 monografie, 2 učebníc, 1 skrípt, 6 kapitol v učebniciach, je autorkou alebo spoluautorkou 56 pôvodných vedeckých prác (z toho 24 s IF), 15 prehľadových prác a zborníkových článkov, s citačným ohlasom 220, z toho 184 citácii je registrovaných v citačných indexoch WOS a SCOPUS. Od roku 2007 je vedeckou sekretárkou Slovenskej neurologickej spoločnosti SLS, je členkou redakčnej rady časopisu Česká a slovenská neurologie a neurochirurgie. V rokoch 2000–2003 a 2007–2010 bola prodekankou LF UPJŠ v Košiciach.
Sources
1. Budka H, Head MW, Ironside JW et al. Sporadic Creutzfeldt-Jakob disease. In: Dickinson DW, Weller RO (eds). Neurodegeneration: the molecular pathology of dementia and movement disorders. 2nd ed. Chichester: Blackwell Publishing 2011.
2. Gajdusek DC, Gibbs J, Alpers M. Experimental transmission of Kuru-like syndrome to chimpanzees. Nature 1966; 209(5025): 794–796.
3. Gibbs CJ jr, Gajdusek DC, Asher DM, Alpers MP, Beck E, Daniel PM et al. Creutzfeldt-Jakob disease (spongiform encephalopathy): transmission to the chimpanzee. Science 1968; 161(3839): 388–389.
4. Prusiner SB. The prion diseases. Brain Pathol 1998; 8(3): 499–513.
5. Mitrová E, Wsólová L, Janáková A. Riziko horizontálneho šírenia Creutzfeldtovej-Jakobovej choroby z pohľadu novších poznatkov. Neurol Prax 2007; 3: 155–158.
6. Aguzzi A, Calella AM. Prions: protein aggregation and infectious diseases. Physiol Rev 2009; 89(4): 1105–1152.
7. Cohen FE, Prusiner SB. Pathologic conformations of prion proteins. Annu Rev Biochem 1998; 67: 793–819.
8. Prusiner SB. Prions. Proc Natl Acad Sci USA 1998; 95(23): 13363–13383.
9. Capellari S, Strammiello R, Saverioni D, Kretzschmar H, Parchi P. Genetic Creutzfeldt-Jakob disease and fatal familial insomnia: insights into phenotypic variability and disease pathogenesis. Acta Neuropathol 2011; 121(1): 21–37.
10. Gambetti P, Kong Q, Zou W, Parchi P, Chen SG. Sporadic and familial CJD: classification and characterisation. Br Med Bull 2003; 66: 213–139.
11. Parchi P, Castellani R, Capellari S, Ghetti B, Young K, Chen SG et al. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol 1996; 39(6): 767–778.
12. Head MW, Ironside JW. Review: Creutzfeldt-Jakob disease: prion protein type, disease phenotype and agent strain. Neuropathol Appl Neurobiol 2012; 38(4): 296–310.
13. Collinge J, Sidle KC, Meads J, Ironside J, Hill AF. Molecular analysis of prion strain variation and aetiology of „new variant“ CJD. Nature 1996; 383(6602): 685–690.
14. Telling GC, Parchi P, DeArmond SJ, Cortelli P, Montagna P, Gabizon R et al. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science 1996; 274(5295): 2079–2082.
15. Appleby BS, Zerr I. Sporadic Creutzfeldt-Jakob disease: changes not only in the brain? Neurology 2012; 79(10): 965–966.
16. Mastrianni J. The genetics of prion diseases. Genet Med 2010; 12(4): 187–195.
17. Ironside JW, Knight RS, Head MW. Iatrogenic Creutzfeldt-Jakob disease. In: Dickinson DW, Weller RO (eds). Neurodegeneration: the molecular pathology of dementia and movement disorders. 2nd ed. Chichester: Blackwell Publishing 2011: 381–386.
18. Rusina R, Matěj R. Prionová onemocnění. Neurol Prax 2012; 13(2): 80–84.
19. Comoy EE, Casalone C, Lescoutra-Etchegaray N, Zanusso G, Freire S, Marcé D et al. Atypical BSE (BASE) transmitted from asymptomatic aging cattle to a primate. PLoS One 2008; 3(8): e3017.
20. Béringue V, Herzog L, Reine F, Le Dur A, Casalone C, Vilotte JL et al. Transmission of atypical bovine prions to mice transgenic for human prion protein. Emerg Infect Dis 2008; 14(12): 1898–1901.
21. Kong Q, Zheng M, Casalone C, Qing L, Huang S, Chakraborty B et al. Evaluation of the human transmission risk of an atypical bovine spongiform encephalopathy prion strain. J Virol 2008; 82(7): 3697–3701.
22. Ward HJ, Everington D, Cousens SN, Smith-Bathgate B, Gillies M, Murray K et al. Risk for sporadic Creutzfeldt-Jakob disease. Ann Neurol 2008; 63(3): 347–354.
23. Collinge J, Palmer MS, Dryden AJ. Genetic predisposition to iatrogenic Creutzfeldt-Jakob disease. Lancet 1991; 337(8755): 1441–1442.
24. Global surveillance, diagnosis, and therapy of human Transmissible Spongiform Encephalopaties: report of a WHO consultation. Geneva, Switzerland, 9–11 February 1998. Available from: http://www.who.int/csr/resources/publications/bse/WHO_EMC_ZDI_98_9/en/.
25. WHO manual for surveillance of human transmissible spongiform encephalopathies including variant Creutzfeldt-Jakob disease. Geneva, Switzerland 2003. Available from: http://whqlibdoc.who.int/publications/2003/9241545887.pdf.
26. Zerr I, Kallenberg K, Summers DM, Romero C, Taratuto A, Heineman U et al. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain 2009; 132(Pt10): 2659–2668.
27. Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 1999; 46(2): 224–233.
28. Parchi P, de Boni L, Saverioni D, Cohen ML, Ferrer I, Gambetti P et al. Consensus classification of human prion disease histotypes allows reliable identification of molecular subtypes: an inter-rater study among surveillance centres in Europe and USA. Acta Neuropathol 2012; 124(4): 517–529.
29. Parchi P, Strammiello R, Notari S, Giese A, Langeveld JP, Ladogana A et al. Incidence and spectrum of sporadic Creutzfeldt-Jakob disease variants with mixed phenotypes and co-occurrence of PrPSc types: an updated classification. Acta Neuropathol 2009; 118(5): 659–671.
30. Cali I, Castellani R, Yuan J, Al-Shekhlee A, Cohen ML, Xiao X et al. Classification of sporadic Creutzfeldt-Jakob disease revisited. Brain 2006; 129(Pt 9): 2266–2277.
31. Puoti G, Bizzi A, Forloni G, Safar JG, Tagliavini F, Gambetti P. Sporadic human prion diseases: molecular insights and diagnosis. Lancet Neurol 2012; 11(7): 618–628.
32. Budka H, Head MW, Ironside JW et al. Sporadic Creutzfeldt-Jakob disease. In: Dickinson DW, Weller RO (eds). Neurodegeneration: the molecular pathology of dementia and movement disorders. 2nd ed. Chichester: Blackwell Publishing 2011: 325–335.
33. Collins SJ, Sanchez-Juan P, Masters CL, Klug GM, van Duijn C, Poleggi A et al. Determinants of diagnostic investigation sensitivities across the clinical spectrum of sporadic Creutzfeldt-Jakob disease. Brain 2006; 129(Pt9): 2278–2287.
34. Meissner B, Kallenberg K, Sanchez-Juan P, Collie D, Summers DM, Almonti S et al. MRI lesion profiles in sporadic Creutzfeldt-Jakob disease. Neurology 2009; 72(23): 1994–2001.
35. Collie DA, Summers DM, Sellar RJ, Ironside JW, Cooper S, Zeidler M et al. Diagnosing variant Creutzfeldt-Jakob disease with the pulvinar sign: MR imaging findings in 86 neuropathologically confirmed cases. AJNR Am J Neuroradiol 2003; 24(8): 1560–1569.
36. Lukic A, Mead S, Rudge P, Collinge J. Comment on validation of diagnostic criteria for variant Creutzfeldt-Jakob disease. Ann Neurol 2011; 69(1): 212.
37. Krasnianski A, Schulz-Schaeffer WJ, Kallenberg K, Meissner B, Collie DA, Roeber S et al. Clinical findings and diagnostic tests in the MV2 subtype of sporadic CJD. Brain 2006; 129(Pt 9): 2288–2296.
38. Krasnianski A, Meissner B, Schulz-Schaeffer WJ, Kallenberg K, Bartl M, Heinemann U et al. Clinical features and diagnosis of the MM2 cortical subtype of sporadic Creutzfeldt-Jakob disease. Arch Neurol 2006; 63(6): 876–880.
39. Sheardova K, Matej R, Rektorova I. Steroid-responsive hyperintense lesions in a patient with Creutzfeldt-Jakob disease. Cesk Slov Neurol N 2010; 73/106(1): 76–79.
40. Seipelt M, Zerr I, Nau R, Mollenhauer B, Kropp S, Steinhoff BJ et al. Hashimotoʼs encephalitis as a differential diagnosis of Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry 1999; 66(2): 172–176.
41. Haïk S, Brandel JP, Sazdovitch V, Delasnerie-Lauprêtre N, Peoch K, Laplanche JL et al. Dementia with Lewy bodies in a neuropathologic series of suspected Creutzfeldt-Jakob disease. Neurology 2000; 55(9): 1401–1404.
42. Ladogana A, Sanchez-Juan P, Mitrová E, Green A, Cuadrado-Corrales N, Sánchez-Valle R et al. Cerebrospinal fluid biomarkers in human genetic transmissible spongiform encephalopathies. J Neurol 2009; 256(10): 1620–1628.
43. Geschwind MD, Martindale J, Miller D, DeArmond SJ, Uyehara-Lock J, Gaskin D et al. Challenging the clinical utility of the 14-3-3 protein for the diagnosis of sporadic Creutzfeldt-Jakob disease. Arch Neurol 2033; 60(6): 813–816.
44. Matěj R, Rusina R, Koukolík F. 5 let činnosti Národní referenční laboratoře lidských prionových onemocnění při Oddělení patologie a molekulární medicíny FTNsP: naše zkušenosti a přehled literatury. Cesk Slov Neurol N 2007; 70/103(6): 637–642.
45. Mitrová E. Contribution of CSF biomarkers to the early diagnosis of human genetic transmissible spongiform encephalopathies. ENJ 2012; 4(1): 9–14.
46. Zerr I, Pocchiari M, Collins S, Brandel JP, de Pedro Cuesta J, Knight RS et al. Analysis of EEG and CSF 14-3-3 proteins as aids to the diagnosis of Creutzfeldt-Jakob disease. Neurology 2000; 55(6): 811–815.
47. Gmitterová K, Heinemann U, Bodemer M, Krasnianski A, Meissner B, Kretzschmar HA et al. 14-3-3 levels in sporadic Creutzfeldt-Jakob disease differ across molecular subtypes. Neurobiol Aging 2009; 30(11): 1842–1850.
48. Chohan G, Pennington C, Mackenzie JM, Andrews M, Everington D, Will RG et al. The role of cerebrospinal fluid 14-3-3 and other proteins in the diagnosis of sporadic Creutzfeldt-Jakob disease in the UK: a 10-year review. J Neurol Neurosurg Psychiatry 2010; 81(11): 1243–1248.
49. Sanchez-Juan P, Green A, Ladogana A, Cuadrado-Corrales N, Sáanchez-Valle R, Mitrováa E et al. CSF tests in the differential diagnosis of Creutzfeldt-Jakob disease. Neurology 2006; 67(4): 637–643.
50. Atarashi R, Satoh K, Sano K, Fuse T, Yamaguchi N, Ishibashi D et al. Ultrasensitive human prion detection in cerebrospinal fluid by real-time quaking-induced conversion. Nat Med 2011; 17(2): 175–178.
51. Steinhoff BJ, Zerr I, Glatting M, Schulz-Schaeffer W, Poser S, Kretzschmar HA. Diagnostic value of periodic complexes in Creutzfeldt-Jakob disease. Ann Neurol 2004; 56(5): 702–708.
52. Richardson EP Jr, Masters CL. The nosology of Creutzfeldt-Jakob disease and conditions related to the accumulation of PrPCJD in the nervous system. Brain Pathol 1996; 5(1): 33–41.
53. Budka H, Aguzzi A, Brown P, Brucher JM, Bugiani O, Gullotta F et al. Neuropathological diagnostic criteria for Creutzfeldt-Jakob disease (CJD) and other human spongiform encephalopaties (prion diseases). Brain Pathol 1995; 5(4): 459–466.
54. Kretzschmar HA, Ironside JW, DeArmond SJ, Tateishi J. Diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Arch Neurol 1996; 53(9): 913–920.
55. Parchi P, Gambetti P, Capellari S. Genetic Creutzfeldt-Jakob disease. In: Dickinson DW, Weller RO (eds). Neurodegeneration: the molecular pathology of dementia and movement disorders. 2nd ed. Chichester: Blackwell Publishing 2011: 336–345.
56. Hsiao K, Meiner Z, Kahana E, Cass C, Kahana I, Avrahami D et al. Mutation of the prion protein in Libyan Jews with Creutzfeldt-Jakob disease. N Engl J Med 1991; 324(16): 1091–1097.
57. Goldfarb LG, Mitrova E, Brown P, Toh BK, Gajdusek DC. Mutation in codon 200 of scrapie amyloid protein gene in two clusters of Creutzfeldt-Jakob disease in Slovakia. Lancet 1990; 336(87): 514–515.
58. Neufeld MY, Josiphov J, Korczyn AD. Demyelinating peripheral neuropathy in Creutzfeldt-Jakob disease. Muscle Nerve 1992; 15(11): 1234–1239.
59. Antoine JC, Laplanche JL, Mosnier JF, Beaudry P, Chatelain J, Michel D. Demyelinating peripheral neuropathy with Creutzfeldt-Jakob disease and mutation at codon 200 of the prion protein gene. Neurology 1996; 46(4): 1123–1127.
60. Hainfellner JA, Parchi P, Kitamoto T, Jarius C, Gambetti P, Budka H. A novel phenotype in familial Creutzfeldt-Jakob disease: prion protein gene E200K mutation coupled with valine at codon 129 and type 2 protease-resistant prion protein. Ann Neurol 1999; 45(6): 812–816.
61. Goldfarb LG, Petersen RB, Tabaton M, Brown P, LeBlanc AC, Montagna P et al. Fatal familial insomnia and familial Creutzfeldt-Jakob disease: disease phenotype determined by a DNA polymorphism. Science 1992; 258(5083): 806–808.
62. Matej R, Kovacs GG, Johanidesova S, Keller J, Matejckova M, Novakova J et al. Genetic Creutzfeldt-Jakob disease with R208H mutation presenting as progressive supranuclear palsy. Mov Disord 2012; 27(4): 476–479.
63. Brown P, Goldfarb LG, Kovanen J, Haltia M, Cathala F, Sulima M et al. Phenotypic characteristics of familial Creutzfeldt-Jakob disease associated with the codon 178Asn PRPNP mutation. Ann Neurol 1992; 31(3): 282–285.
64. Rusina R, Fiala J, Holada K, Matějčková M, Nováková J, Ampapa R et al. Gerstmann-Sträussler-Scheinker syndrome with the P102L pathogenic mutation presenting as familial Creutzfeldt-Jakob disease: a case report and review of the literature. Neurocase 2013; 19(1): 41–53.
65. Ironside JW, Head MW, Will RG. Variant Creutzfeldt-Jakob disease. In: Dickinson DW, Weller RO (eds). Neurodegeneration: the molecular pathology of dementia and movement disorders. 2nd ed. Chichester: Blackwell Publishing 2011: 354–363.
66. Will RG, Ironside JW, Zeidler M, Cousens SN, Estibeiro K, Alperovitch A et al. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet 1996; 347(9006): 921–925.
67. Llewelyn CA, Hewitt PE, Knight RS, Amar K, Cousens S, Mackenzie J et al. Possible transmission of variant Creutzfeldt-Jakob disease by blood transfusion. Lancet 2004; 363(9407): 417–421.
68. Wroe SJ, Pal S, Siddique D, Hyare H, Macfarlane R, Joiner S et al. Clinical presentation and pre-mortem diagnosis of variant Creutzfeldt-Jakob disease associated with blood transfusion: a case report. Lancet 2006; 368(9552): 2061–2067.
69. Mitrová E. Creutzfeldtova-Jakobova choroba: riziká, výskyt a možnosti diagnostiky najvýznamnejšej prenosnej demencie. Neurol Prax 2004; 1: 29–32.
70. World Health Organization. The revision of the surveillance case definition for variant Creutzfeldt-Jakob disease (vCJD). Available from: http://whqlibdoc.who.int/hq/2002/WHO_CDS_CSR_EPH_2001.5.pdf.
71. Will RG, Zeidler M, Stewart GE, Macleod MA, Ironside JW, Cousens SN et al. Diagnosis of new variant Creutzfeldt-Jakob disease. Ann Neurol 2000; 47(5): 575–582.
72. Summers DM, Collie DA, Zeidler M, Will RG. The pulvinar sign in variant Creutzfeldt-Jakob disease. Arch Neurol 2004; 61(3): 446–447.
73. Yamada M, Variant CJD Working Group, Creutzfeldt-Jakob Disease Surveillance Committee, Japan. The first Japanese case of variant Creutzfeldt-Jakob disease showing periodic elektroencephalogram. Lancet 2006; 367(9513): 874.
74. Binelli S, Agazzi P, Giaccone G, Will RG, Bugiani O, Franceschetti S et al. Periodic elektroencephalogram complexes in a patient with variant Creutzfeldt-Jakob disease. Ann Neurol 2006, 59(2): 423–427.
75. Head MW, Ritchie D, Smith N, McLoughlin V, Nailon W, Samad S et al. Peripheral tissue involvement in sporadic, iatrogenic and variant Creutzfeldt-Jakob disease: an immunohistochemical, quantitative and biochemical study. Am J Pathol 2004; 164(1): 143–153.
76. Brown P, Brandel JP, Preece M, Sato T. Iatrogenic Creutzfeldt-Jakob disease: the waning of an era. Neurology 2006; 67(8): 389–393.
77. Jirsova K, Krabcova I, Novakova J, Hnathova I, Koukolik F, Kubesova B et al. The assessment of pathogenic prions in the brains of eye tissue donors: 2-years experience in the Czech Republic. Cornea 2010; 29(9): 996–999.
78. Mitrova E, Cernak A, Slivarichova D, Koscova S, Bernovska V, Cernak M. Experience with preventive genetic testing of corneal donors in Slovakia. Cornea 2011; 30(9): 987–990.
79. Heath CA, Barker RA, Esmonde TF, Harvey P, Roberts R, Trend P et al. Dura mater-associated Creutzfeldt-Jakob disease: experience from surveillance in the UK. J Neurol Neurosurg Psychiatry 2006; 77(7): 880–882.
80. Brown P, Preece M, Brandel JP, Sato T, McShane L, Zerr I et al. Iatrogenic Creutzfeldt-Jakob disease at the millennium. Neurology 2000; 55(8): 1075–1081.
81. Geschwind MD, Shu H, Haman A, Sejvar JJ, Miller BL. Rapidly progressive dementia. Ann Neurol 2008; 64(1): 97–108.
82. Vernino S, Geschwind M, Boeve B. Autoimmune encephalopathies. Neurologist 2007; 13(3): 140–147.
83. Lawn ND, Westmoreland BF, Kiely MJ, Lennon VA, Vernino S. Clinical, magnetic resonance imaging, and electroencephalographic findings in paraneoplastic limbic encephalitis. Mayo Clinic Proc 2003; 78(11): 1363–1368.
84. Doležalová I, Kuba R, Musilová K, Kašpárek T, Brázdil M, Rektor I. Neparaneoplastická limbická encefalitida s pozitivitou anti-LGI1 protilátek. Neurol Prax 2012; 13(3): 221–224.
85. Zborníková P, Krasulová E, Bušek P, Nytrová P, Peterová V, Špačková N et al. Autoimunitní limbická encefalitida asociovaná s LGI1 protilátkami. Neurol Prax 2012; 13(6): 350–353.
86. Thieben MJ, Lennon VA, Boeve BF, Aksamit AJ, Keegan M, Vernino S. Potentially reversible autoimmune limbic encephalitis with neuronal potassium channel antibody. Neurology 2004; 62(7): 1177–1182.
87. Vincent A, Buckley C, Schott JM, Baker I, Dewar BK, Detert N et al. Potassium channel antibody-associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis. Brain 2004; 127(Pt 3): 701–712.
88. Dalmau J, Tüzün E, Wu HY, Masjuan J, Rossi JE, Voloschin A et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol 2007; 61(1): 25–36.
89. Castillo P, Woodruff B, Caselli R, Vemino S, Lucchinetti C, Swanson J et al. Steroid-responsive encephalopathy associated with autoimmune thyroiditis. Arch Neurol 2006; 63(2): 197–202.
90. Salvarani C, Brown RD Jr, Calamia KT, Christianson TJ, Weigand SD, Miller DV et al. Primary central nervous system vasculitis: analysis of 101 patients. Ann Neurol 2007; 62(5): 442–451.
91. Amara AW, Bashir K, Palmer CA, Walker HC. Challenges in diagnosis of isolated central nervous system vasculitis. Brain Behav 2011; 1(1): 57–61.
92. Bushara KO. Neurologic presentation of celiac disease. Gastroenterology 2005; 128 (4 Suppl 1): S92–S97.
93. Itzhaki RF, Wozniak MA. Viral infection and cognitive decline. J Am Geriatr Soc 2007; 55(1): 131.
94. Almeida OP, Lautenschlager NT. Dementia associated with infectious diseases. Int Psychogeriatr 2005; 17 (Suppl 1): S65–S77.
95. Timmermans M, Carr J. Neurosyphylis in the modern era. J Neurol Neurosurg Psychiatry 2004; 75(12): 1727–1730.
96. Fallon BA, Keilp JG, Corbera KM, Petkova E, Britton CB, Dwyer E et al. A randomized, placebo-controlled trial of repeated IV antibiotic therapy for Lyme encephalopathy. Neurology 2008; 70(13): 992–1003.
97. Kennedy PG. Human African trypanosomiasis-neurological aspects. J Neurol 2006; 253(7): 411–416.
98. Josephson SA, Papanastassiou AM, Berger MS, Barbaro NM, McDermott MW, Hilton JF et al. The diagnostic utility of brain biopsy procedures in patients with rapidly deteriorating neurological conditions or dementia. J Neurosurg 2007; 106(1): 72–75.
99. Lai R, Rosenblum MK, DeAngelis LM. Primary CNS lymphoma: a whole-brain disease? Neurology 2002; 59(10): 1557–1562.
100. Menendez Calderon MJ, Segui Riesco ME, Argüelles M, Nuño Mateo J. Intravascular lymphomatosis. A report of three cases. An Med Interna 2005; 22(1): 31–34.
101. Schott JM, Reiniger L, Thom M, Holton JL, Grieve J, Brandner S et al. Brain biopsy in dementia: clinical indications and diagnostic approach. Acta Neuropathol 2010; 120(3): 327–341.
102. Kratochvílová J. Doporučený pracovní postup pro ošetřování pacientů s podezřením na CJN, nvCJN a režim dekontaminace a sterilizace. Available from: www.sneh/cz/.
103. Koukolík F. Pitvy a bioptická vyšetření mozku při podezření na prionové choroby. Available from: http://www.patologie.info/soubor/standardy/11-Pitvy_a_biopticka_vysetreni_mozku_pri_podezreni_na_prionove_choroby.doc.
104. Mitrová E. Transmissibilné spongioformné encefalopatie. Prionové choroby. Budapešť Press Publica 1999.
105. Zerr I. Therapeutic trials in human transmissible spongiform encephalopaties: recent advances and problems to address. Infect Disord Drug Targets 2009; 9(1): 92–99.
106. Collins SJ, Lewis V, Brazier M, Hill AF, Fletcher A, Masters CL. Quinacrine does not prolong survival in a murine Creutzfeldt-Jakob disease model. Ann Neurol 2002; 52(4): 503–506.
107. Otto M, Cepek L, Ratzka P, Doehlinger S, Boekhoff I, Wiltfang J et al. Efficacy of flupirtine on cognitive function in patients with CJD: a double-blind study. Neurology 2004; 62(5): 714–718.
108. Forloni G, Salmona M, Marcon G, Tagliavini F. Tetracyclines and prion infectivity. Inf Disord Drug Targets 2009; 9(1): 23–30.
109. Rusina R, Rusinová K, Holmerová I, Šimek J. Léčba pokročilé demence – paliativní přístup. Neurol Prax 2010; 11: 16–19.
110. Koukolík F. Lidská prionová onemocnění jsou v ČR pod kontrolou už 10 let. Available from: www.ftn.cz
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2013 Issue 2
Most read in this issue
- Creutzfeldtova-Jakobova choroba
- Spinocerebelární ataxie typ 7 (SCA7) – kazuistika
- Lymeská borelióza jako příčina bilaterální neuroretinitidy s výraznou jednostrannou hvězdicovitou makulopatií u osmileté dívky
- Elektrofyziologické vyšetření pánevního dna