
Spinální komplikace u genetických syndromů – kazuistiky
Spinal Complications in Genetic Syndromes – Case Reports
Distinct genetic syndromes (achondroplasia, Down’s syndrome) are associated with congenital or degenerative vertebral abnormalities and with involvement of the ligamentous apparatus. In patients with achondroplasia, vertebral deformities may result in spinal stenosis, nerve root compression and development of myelopathy. The most common spinal complication in Down’s syndrome is atlanto-axial instability. This condition is associated with a shift of C1/C2 vertebrae and subsequent compression of dorsal roots, spinal cord and the development of cervical myelopathy. Two case reports of patients with these genetic disorders are presented. Surgical intervention was considered necessary in both of them.
Key words:
achondroplasia – Down´s syndrome – spinal stenosis – atlantoaxial instability – spinal fusion – decompression laminectomy
Authors:
M. Jakubíková 1; I. Příhodová 1; M. Barna 2; J. Štulík 2; P. Vaněk 3; V. Beneš; S. Nevšímalová 1
Authors‘ workplace:
Neurologická klinika a Centrum klinických neurověd 1. LF UK a VFN v Praze
1; Spondylochirurgické oddělení III. chirurgické kliniky 1. LF UK a FN v Motole, Praha
2; Neurochirurgická klinika 1. LF UK a ÚVN Praha
3
Published in:
Cesk Slov Neurol N 2011; 74/107(4): 482-485
Category:
Case Report
Overview
Součástí některých genetických syndromů (achondroplazie, Downův syndrom) jsou anomálie či degenerativní změny obratlů a odchylky ligamentózního aparátu. Strukturální změny obratlových těl s následnou spinální stenózou vedou u pacientů s achondroplazií k útlaku nervových kořenů a někdy i k rozvoji myelopatie. Nejčastější spinální komplikací pacientů s Downovým syndromem je tzv. atlantoaxiální instabilita. Může docházet k posunu obratlů C1/C2 s následným útlakem míšních kořenů, míchy a rozvojem cervikální myelopatie. Uvádíme dvě kazuistiky pacientů s touto problematikou. U obou pacientů byla nutná chirurgická intervence.
Klíčová slova:
achondroplazie – Downův syndrom – spinální stenóza – atlantoaxiální instabilita – spinální fúze – dekompresní laminektomie
Úvod
Achondroplazie a Downův syndrom jsou geneticky podmíněná onemocnění spojená s anomáliemi a degenerativními změnami obratlů. Tyto odchylky mají za následek zvýšené riziko rozvoje neurologických příznaků způsobených kompresí nervových kořenů a míchy.
Achondroplazie je autozomálně dominantně dědičné onemocnění pohybového aparátu podmíněné poruchou receptorů odpovídajících za vazbu růstového faktoru fibroblastů. Incidence tohoto onemocnění je 1 : 15 000 až 40 000 živě narozených dětí [1]. Tito pacienti mají typicky malý vzrůst s krátkými končetinami, lumbální hyperlordózu, valgózní nebo varózní postavení dolních končetin [2].
Popisovány jsou rovněž abnormality axiálního skeletu jako deformity baze lební s úzkým foramen magnum a nízká obratlová těla s menšími pedikly. V důsledku menších pediklů a snižujících se interpedikulárních vzdáleností je kongenitálně úzký spinální kanál, který se věkem, vlivem degenerativních změn ještě více zužuje a navozuje neurologické potíže [3].
Achondroplazie je sdružena s neurologickými jednotkami jako hydrocefalus, komprese, cervikální nebo torakální míšní komprese, lumbální spinální stenóza [2,4]. U Downova syndromu je poměrně častá atlantoaxiální instabilita, která se vyskytuje u 9,5–27 % pacientů [5], u 1–2 % je symptomatická. Příčinou je laxicita nebo špatný vývin ligament, anomálie C2 obratle a časté degenerativní změny obratlů. Dochází tak k posunu obratlů C1/C2 s následným útlakem míšních kořenů, míchy a rozvojem cervikální myelopatie [6]. Atlantoaxiální instabilita je forma cervikální subluxace, kterou diagnostikujeme převážně u pacientů s trizomií 21. chromozomu.
Kazuistika 1
Sedmnáctiletý pacient s diagnózou achondroplazie (potvrzena mutace v genu FGFR3 – fibroblast growth factor receptor – v heterozygotním stavu) byl akutně přijat pro dva týdny trvající zhoršování chůze a slabost dolních končetin s levostrannou převahou. V anamnéze byl údaj o evakuaci hygromu bifrontálně ve věku dvou let pro nárůst velikosti subdurálního prostoru a výrazné oboustranné rozšíření subarachnoidálních prostor. Měl dlouhodobě bolesti zad, byl dispenzarizován na rehabilitaci, neurologii a ortopedii. Při přijetí si stěžoval kromě slabosti dolních končetin také na bolesti v bederní páteři s propagací do obou dolních končetin po zevní straně ke kolenům. Udával postupné zhoršování citlivosti na dolních končetinách od třísel dolů, sfinkterové obtíže negoval.
V objektivním neurologickém nálezu byla při příjmu periferní paraparéza dolních končetin, více vlevo s maximem v segmentech L2–L4 a taktilní porucha čití v dermatomech L4 a L5. V den přijetí došlo k jednorázovému výpadku sfinkterů.
Statimové vyšetření magnetickou rezonancí (MR) lumbosakrální (LS) páteře bylo pro četné pohybové artefakty obtížně hodnotitelné, patrné bylo více etážové zúžení konstitučně již velmi úzkého páteřního kanálu.
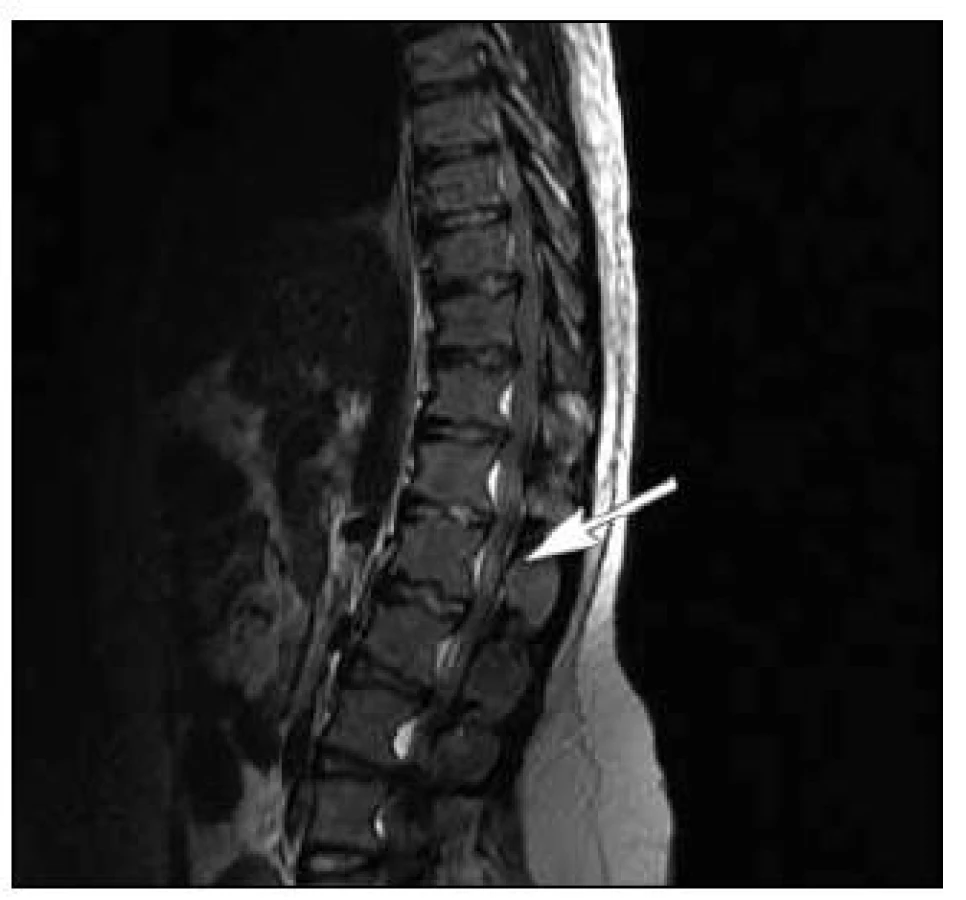
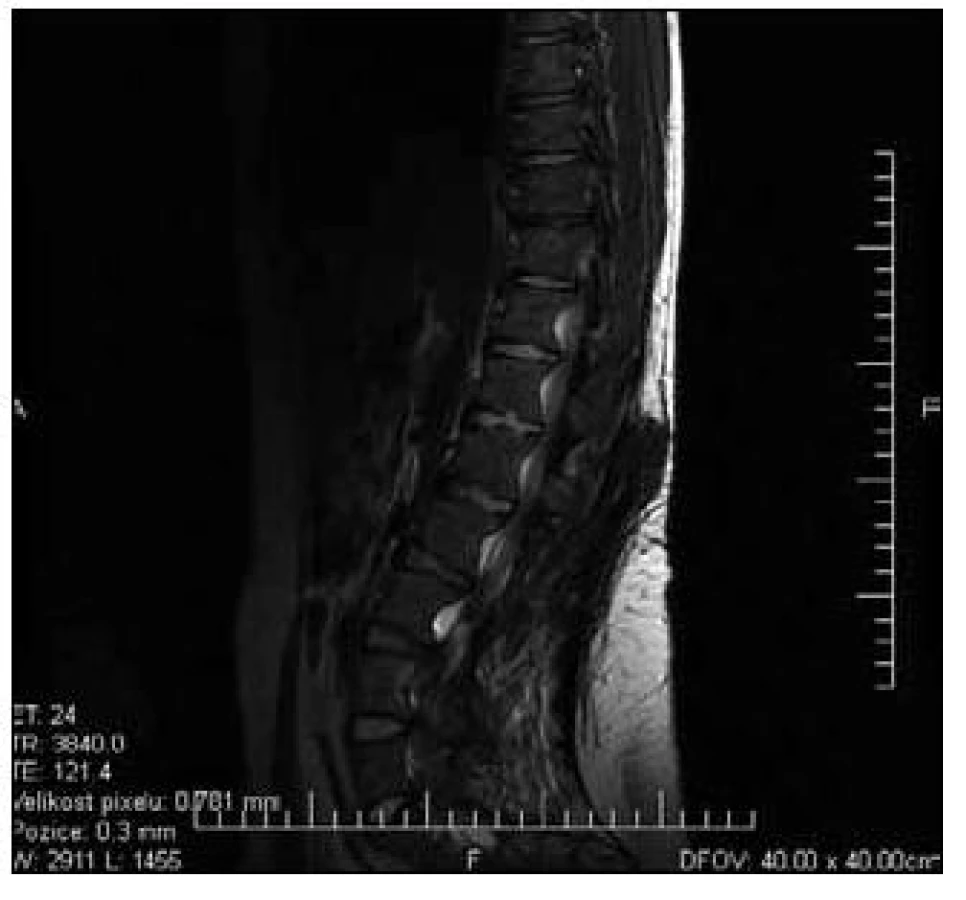
Pacient byl přeložen na Neurochirurgickou kliniku ÚVN, kde byla týž den provedena MR LS páteře v celkové anestezii. Nalezena byla typická achondroplastická páteř s polysegmentální stenózou hrudního a bederního úseku s maximem v úrovni L2/L3 až L4/L5, kde byla patrna absolutní stenóza páteřního kanálu se zúžením foramin oboustranně (obr. 1). Maximum stenózy bylo verifikováno i perimyelograficky. Druhý den po přijetí byla provedena dekompresní laminektomie v úseku L2–L5 (obr. 2). Neurochirurgický výkon proběhl bez komplikací. Pooperační průběh byl rovněž klidný, pacient rehabilitoval, postupně se vertikalizoval, za tři týdny od výkonu byl schopen samostatné chůze, porucha taktilního čití se výrazně zlepšila, sfinktery byly intaktní.
Kazuistika 2
Devítiletá pacientka z II. fyziologické gravidity, narozena předčasně v 36. týdnu, nekříšena, měla slabý ikterus. Postnatálně byla nápadná genetická stigmatizace a hypotonie, diagnostikován Downův syndrom (potvrzena trizomie 21. chromozomu). Posléze byla zjištěna srdeční vada charakteru atrioventrikulárního kanálu s úplným defektem atrioventrikulárního septa, pulmonální valvulární stenóza a trikuspidální insuficience. V osmi měsících věku byla provedena kardiochirurgická intervence s výborným efektem. Pacientka měla výrazně opožděný psychomotorický vývoj, mentální úroveň se pohybovala v pásmu středně těžké mentální retardace. Od kojeneckého věku byla umístěna v ústavu sociální péče.
Na naší kliniku byla přijata pro půl roku trvající a postupně progredující poruchu chůze, častější pády, dominantní byla slabost pravostranných končetin.
V objektivním neurologickém nálezu byla kvadrupyramidová symptomatologie s pravostrannou převahou, hypotonický syndrom, mentální retardace a typická stigmatizace při základní genetické vadě. Pro slabost dolních končetin již nebyla schopna chůze.
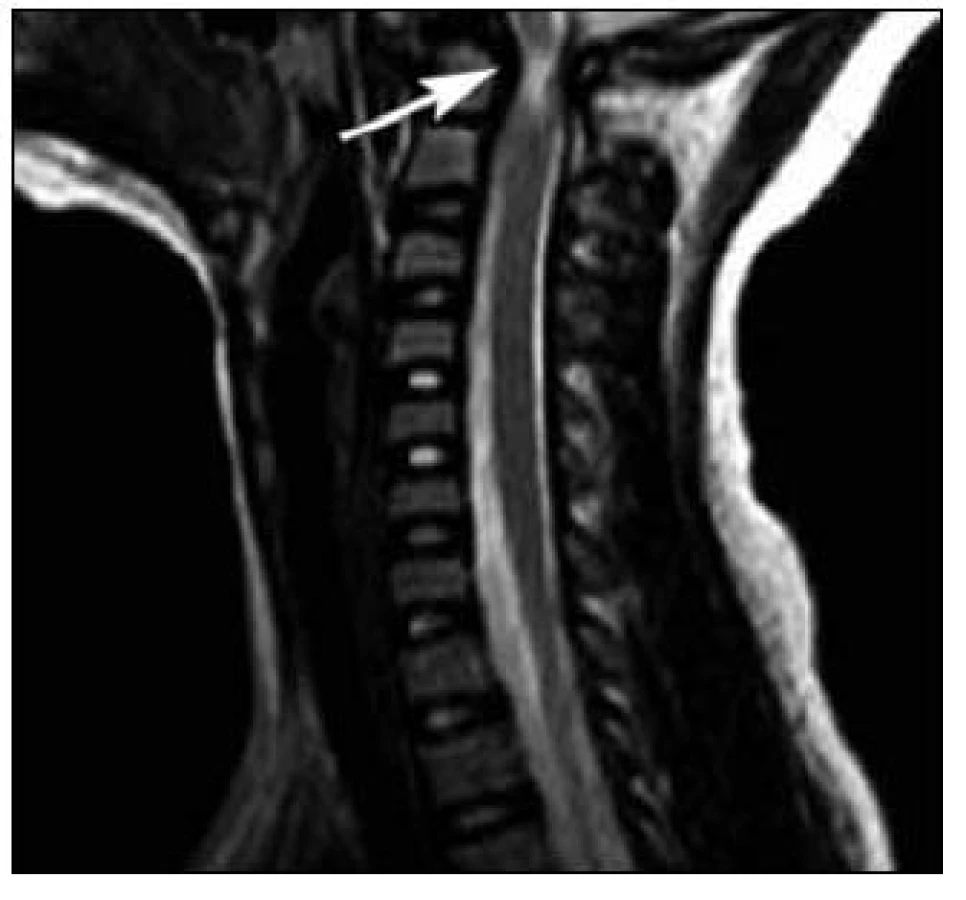
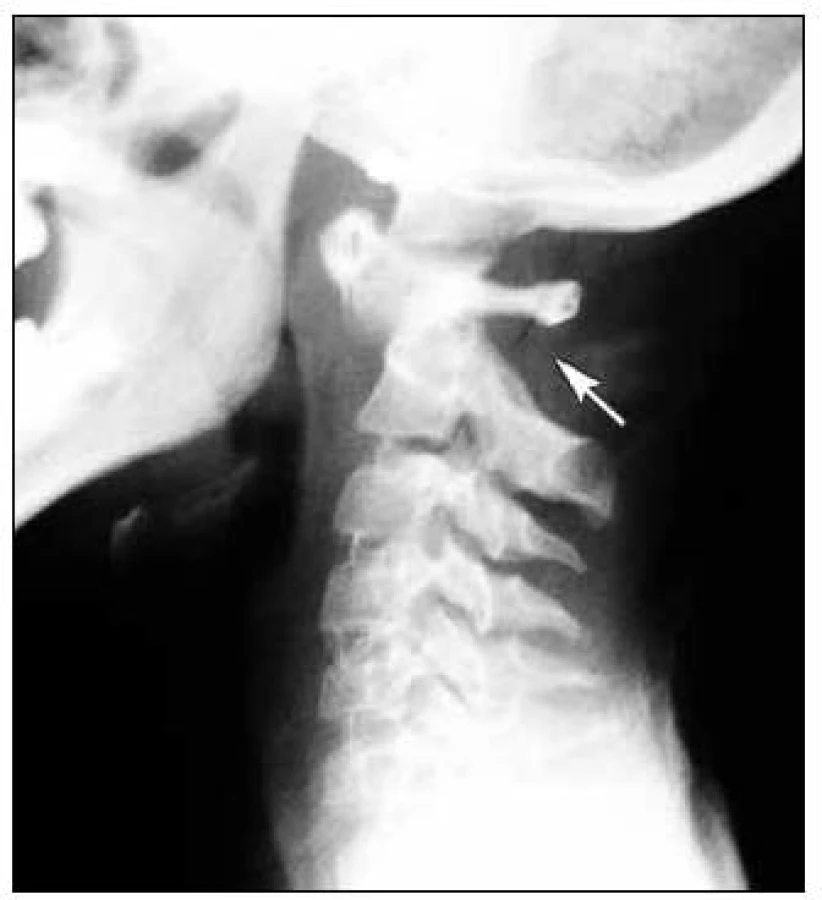
MR mozku a krční páteře prokázalo destrukci dens axis s jeho přeměnou charakteru pannu s následnou prominencí do páteřního kanálu. Nález způsoboval těsnou spinální stenózu v tomto segmentu s rozsáhlou myelopatií (obr. 3, 4). Pacientka byla přeložena na spondylochirurgické oddělení FN v Motole a tam byla provedena spondylochirurgická repozice a zadní stabilizace krční páteře v úseku C1–C2 dle Harmse (obr. 5, 6).
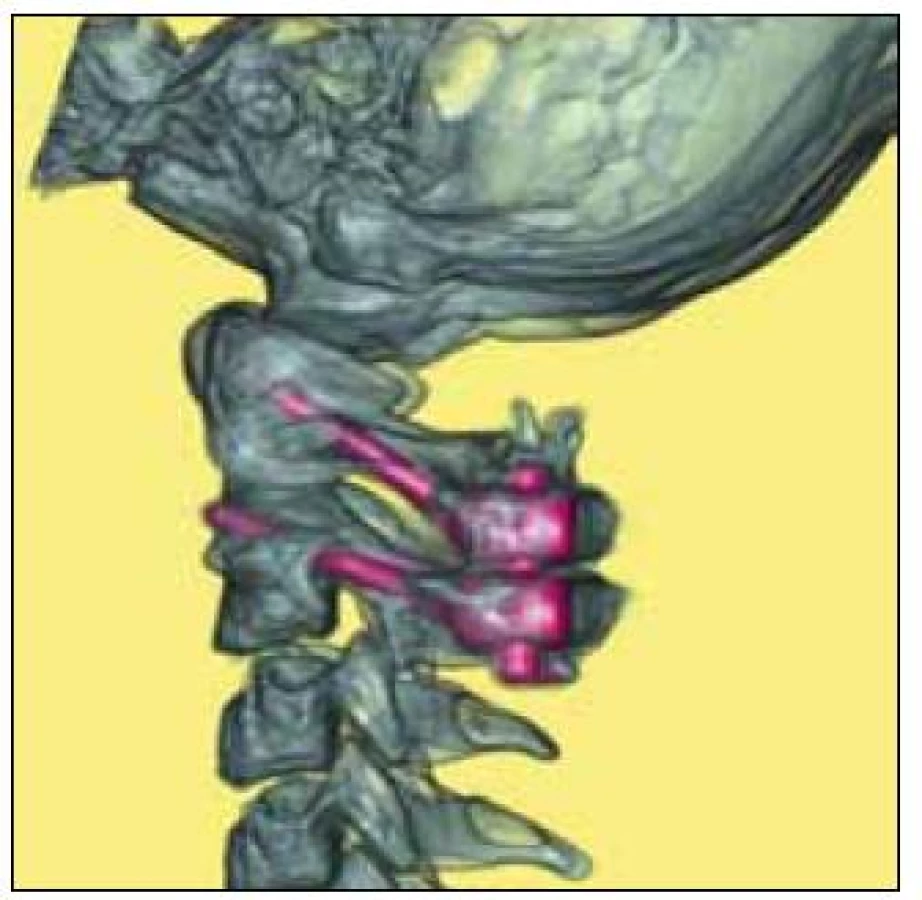

Pooperační průběh byl bez komplikací, došlo k výraznému zlepšení neurologického nálezu a po šesti týdnech od výkonu byla pacientka schopna chůze s oporou.
Diskuze
U achondroplazie je molekulárně genetickou podstatou onemocnění bodová mutace v transmembránové doméně genu FGFR-3, který se nachází na dlouhém raménku 4. chromozomu, což vede k vzniku defektních receptorů pro růstový faktor fibroblastů [7].
Termín achondroplasia, který znamená nedostatečné chrupavčité utváření, byl poprvé použit Parrotem v roce 1878 [8]. Toto onemocnění je nejčastější typ tzv. disproporčního trpaslictví, kde primární porucha je v abnormální enchondrální osifikaci. Dochází proto k rizomelickému zkrácení končetin a současně k patologickému utváření axiálního skeletu. V důsledku úzkého foramen magnum a zvýšeného intrakraniálního žilního tlaku může vzniknout komunikující hydrocefalus nebo při stenóze akvaduktu hydrocefalus nekomunikující [9]. U našeho pacienta bylo již od kojeneckého věku pozorováno rozšíření subarachnoidálních prostor, které je rovněž typické pro tuto diagnózu. Následně byla provedena evakuace oboustranného subdurálního hygromu.
Autoři Donath a Vogl již v roce 1925 popsali strukturální změny obratlových těl, které jsou odpovědné za další závažné neurologické komplikace této choroby – myelopatie a útlaky nervových kořenů [10]. Spinální stenózy a stenózy intervertebrálních foramen jsou u pacientů s diagnózou achondroplazie častým jevem. Někdy jsou přítomny známky spinální stenózy od narození, častěji se však projevy spinální nebo radikulární komprese objevují až ve středním nebo vyšším věku. Přibližně u 1/3 nemocných se tato symptomatika vyvine před 15. rokem, v některých studiích [1] měla více než polovina pacientů příznaky dekompenzované spinální stenózy již od 12 let věku. Odhaduje se, že asi 10–25 % pacientů všech věkových kategorií s touto diagnózou postupně dospěje k neurochirurgické léčbě [11].
U našeho pacienta se jednalo o chronické obtíže charakteru iritačního radikulárního syndromu L5 bilaterálně, které dva týdny před přijetím prudce progredovaly do obrazu akutního syndromu kaudy (slabosti dolních končetin s nemožností chůze provázené poruchou taktilního čití a incipientními sfinkterovými obtížemi).
Zobrazovací vyšetření prokázala kongenitálně úzký spinální kanál s dekompenzací a útlakem kaudy v oblasti L2–L5, které si vyžádalo akutní neurochirurgickou intervenci v podobě rozsáhlé laminektomie.
Podobně jako u achondroplazie i u Downova syndromu neurologická symptomatika souvisí s mechanickou kompresí buď na úrovni nervových kořenů nebo míchy s následným vývojem myelopatie. Nejzávažnější a někdy i život ohrožující komplikací je cervikální myelopatie s následným a často plíživým rozvojem spastické paraparézy dolních končetin, vývojem kvadruparézy nebo vzácněji hemiparézy. Problém však je, že někdy spasticita i při těžké cervikální myelopatii nemusí být nápadná díky hypotonickému syndromu, který je u Downova syndromu zpravidla přítomen. Nepřímé klinické známky, jež mohou být asociovány s atlantoaxiální instabilitou, jsou např. změny v chování, vyhýbání se jistým aktivitám, změna preference ruky nebo pomočování u dítěte, které již bylo dlouhodobě kontinentní [6].
U naší pacientky byl vývin symptomů pomalý a plíživý, klinický obraz postupně progredoval až do obrazu kvadruparézy s pravostrannou převahou, byla přítomna spasticita s iritačními pyramidovými jevy. Sfinktery nebylo možné hodnotit pro přetrvávající inkontinenci při mentální retardaci.
Nejčastější příčinou atlantoaxiální instability je vrozená odchylka ligamentózního aparátu neboli laxicita ligamentum transversum atlantis, které přitlačuje dens axis proti ventrálnímu oblouku atlasu. Výskyt u Downova syndromu poprvé popsali v roce 1960 Spitzer et al [12]. Příčinou (kromě patologie na úrovni ligament) může být i vrozená malformace atlasu [6].
Atlantoaxiální instabilita u Downova syndromu může být zcela asymptomatická a odhalí ji až náhodný RTG nález. Vzhledem k nenápadnému klinickému obrazu, který dokáže ujít pozornosti, je u všech pacientů s tímto syndromem doporučován skríning nativním RTG – bočný dynamický RTG snímek (flexe, normální pozice, extenze). Pokud je atlantoodontoideální vzdálenost při flexi větší než 3 mm, je doporučováno každoroční sledování včetně neurologického vyšetření. Až 17 % asymptomatických dětí s Downovým syndromem má atlantoodontoideální vzdálenost větší než 5 mm, u symptomatických pacientů s tímto nálezem je vždy indikováno MR krční páteře. Vzdálenost nad 6 mm se považuje za radiologickou známku atlantoaxiální instability [6].
Závěr
Neurologické příznaky spojené s kompresí nervových kořenů a míchy se u pacientů s Downovým syndromem a achondroplazií vyskytují častěji v důsledku anomálií jejich skeletálního systému. Zejména u pacientů s Downovým syndromem může jejich rozvoj ujít pozornosti vzhledem k mentální retardaci a pozvolnému průběhu. Je vhodný pravidelný skríning zobrazovacími metodami i u asymptomatických pacientů.
Podpořeno výzkumným záměrem MSM 0021620849
MUDr. Michala Jakubíková
Neurologická
klinika a Centrum klinických neurověd
1. LF UK a VFN
Kateřinská
30
128
21 Praha 2
e-mail:
michala.jakubikova@vfn.cz
Přijato
k recenzi: 29. 10. 2010
Přijato
do tisku: 6. 1. 2011
Sources
1. Sciubba D, Noggle J, Marupudi N, Bagley C, Bookland M, Carson B et al. Spinal stenosis surgery in pediatric patients with achondroplasia. J Neurosurgery 2007; 106 (Suppl 5): 372–378.
2. Ain MC, Browne JA. Spinal arthrodesis with instrumentation for thoracolumbar kyphosis in pediatric achondroplasia. Spine 2004; 29(18): 2075–2080.
3. Jeong ST, Song HR, Keny SM, Telang SS, Suh SW, Hong SJ. MRI study of the lumbar spine in achondroplasia: a morphometric analysis for the evaluation of stenosis of the canal. J Bone Point Surg Br 2006; 88(9): 1192–1196.
4. Bagley CA, Pindrik JA, Bookland MJ, Camara-Quintana JQ, Carson BS. Cervicomedullary decompression for foramen magnum stenosis in achondroplasia. J Neurosurg 2006; 104 (Suppl 3): 166–172.
5. Pueschel SM, Scola FH. Epidemiological radiographic and clinical studies of atlantoaxial instability in individuals with Down’s syndrome. Pediatrics 1987; 80 : 555–560.
6. Alvarez N, Rubin L. Atlantoaxial instability in adults with down syndrome: a clinical and radiological survey. Applied Research in Mental Retardation 1986; 1(7): 67–78.
7. Baitner AC, Maurer SG, Gruener MB, Di Cesare PE. The genetic basis of the osteochondrodysplasias. J Pediatr Orthop 2000; 20(5): 594–605.
8. Parrot JM. Les malformations achondrodysplasiques. Bulletines de la Société d’antropologie de Paris 1878.
9. Gordon N. The neurological complications of achondroplasia. Brain Dev 2000; 22(1): 3–7.
10. Donath J, Vogl A. Untersuchungen uber den chondrodystrophischen Zwergwuchs. Wien Arch Inn Med 1925; 10 : 1–44.
11. Hall JG. The natural history of achondroplasia. Basic Life Sci 1988; 48 : 3–9.
12. Spitzer R, Rabinovitch JY, Wyber KC. A study of abnormalities of the skull, teeth and lenses in mongolism. Can Med Assoc J 1961; 84 : 567–572.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery
2011 Issue 4
Most read in this issue
- Porucha pozornosti s hyperaktivitou (attention deficit/hyperactivity disorder – ADHD)
- Opožděný akutní subdurální hematom
- Neurologické komplikace při onemocnění herpes zoster – kazuistika
- Vrozená myotonie na podkladě mutací v genu pro chloridový kanál ClC-1