
Hemimegalencefalie: přehled literatury a zkušenosti s chirurgickou léčbou 5 nemocných dětí
Hemimegalencephalia. An overview of relevant literature and experience in surgical treatment of 5 affected children
Hemimegalencephalia is a dysplasia deforming the cerebral cortex and causing asymmetry of the cerebral hemispheres. The dominating feature of an MR image is the abnormal size of one of the hemispheres, ventriculomegaly and abnormality of gyrification. The disease can exist in isolation or in connection with other lesions forming part of a neurocutaneous disease. The three-grade classification (I–III) reflects the severity of affection and prognosis. The disease is manifested as early as in childhood, mostly in the form of refractory epileptic seizures and other psychological and neurological symptoms. If conservative treatment fails, surgical removal of the affected area is the method of choice: lobar/multilobar resection (A), subpial transsection in the motor area (B), decortication (C), and functional or anatomic hemispherectomy (fHSE or aHSE, respectively). The authors report the course and results of surgical treatment of five children with the hemimegalencephalia diagnosis. The clinical picture of all the five children was dominated by refractory epileptic seizures, psycho-motor development disorder and additional neurologic symptoms: contralateral hemiparesis, myoclonal jerks etc. Surgery was performed in all the patients: calosotomy and functional hemispherectomy extended in the second time, subpial transsection of the motor area with multilobar resection, subpial transsection of the motor area with lobar resection and decortication, anatomic hemispherectomy, functional hemispherectomy in the second time complemented with anatomic hemispherectomy. In all cases, the treatment resulted in lower frequency of EP seizures, and the progress of PM slightly decelerated in three patients, while the surgery had no effect in another two patients with stagnating PM development. Thirty-day morbidity and mortality in the set was nil. The authors refer to hemimegalencephalia as a relatively rare neurosurgical disease. They point out that also the relatively invasive surgery which is only sporadically used today, has its place in the treatment of hemimegalencephalia.
Key words:
cerebral cortex dysplasia – hemimegalencephalia – refractory epilepsy – hemispherectomy
Authors:
H. Homolková 1; M. Häckel 2; M. Prchlík 1; V. Beneš 2
Authors‘ workplace:
Klinika dětské chirurgie a traumatologie 3. LF UK a FTN Praha
1; Neurochirurgická klinika 1. LF UK a ÚVN Praha a IPVZ
2
Published in:
Cesk Slov Neurol N 2007; 70/103(5): 538-543
Category:
Short Communication
Overview
Hemimegalencefalie patří mezi dysplazie deformující mozkovou kůru, způsobuje asymetrii mozkových hemisfér. V MRI-obraze dominuje abnormální velikost jedné hemisféry, ventrikulomegalie a porucha gyrifikace. Může se vyskytovat jako izolované onemocnění nebo ve spojení s jinými lézemi v rámci neurokutánního onemocnění. Třístupňová klasifikace (I–III) zohledňuje tíži postižení a prognózu. Onemocnění se manifestuje již v dětském věku převážně refrakterními epileptickými záchvaty a dalšími psychologickými a neurologickými příznaky. Při selhání možností konzervativní léčby je metodou volby chirurgické odstranění postižené oblasti: lobární/multilobární resekce (A), subpiální transsekce v motorické krajině (B), dekortikace (C), a funkční (fHSE) nebo anatomická hemisferektomie (aHSE). Autoři referují průběh a výsledky chirurgické léčby pěti dětí s diagnózou hemimegalencefalie. V klinickém obraze všech nemocných dětí dominovaly refrakterní epileptické záchvaty, porucha psychomotorického vývoje a přítomny byly další neurologické příznaky: kontralaterální hemiparéza, myoklonické záškuby aj. U všech nemocných byla provedena chirurgická léčba: kalozotomie a v druhé době rozšířená funkční hemisférektomie, subpiální transsekce motorické krajiny s multilobární resekcí, subpiální transsekce motorické krajiny s lobární resekcí a dekortikací, anatomická hemisférektomie, funkční hemisférektomie v druhé době doplněna anatomickou hemisférektomií. Výsledky léčby přinesly ve všech případech snížení četnosti EP záchvatů, u tří nemocných došlo k lehkému zpomalení PM vývoje a u dvou nemocných se stagnací PM vývoje nepřinesl chirurgický zákrok v PM vývoji žádnou změnu. 30denní morbidita a mortalita souboru je nulová. Autoři připomínají hemimegalencefalii jako nepříliš časté neurochirurgické onemocnění. Zdůrazňují, že i relativně invazivní, dnes sporadicky užívaná chirurgická metoda, hemisférektomie má své místo při léčbě hemimegalencefalie.
Klíčová slova:
dysplazie mozkové kůry – hemimegalencefalie – refrakterní epilepsie – hemisférektomie
Úvod
V posledních 15 letech došlo na poli výzkumu patogeneze některých vrozených vývojových vad k významným objevům, zejména na molekulární a genetické úrovni [1–3]. Další pokrok ve vývoji zobrazovacích technik, zejména kvalita a dostupnost MRI, rozšířil diagnostické možnosti vrozených vad a umožnil získat přesnější a podrobnější představy o vzácnějších nebo dosud málo známých onemocněních [4].
Hemimegalencefalie (dále jen HME) patří k řidčeji vídaným asymetrizujícím malformacím mozku ze skupiny hamartomatóz. HME se vyskytuje v izolované formě nebo sdružená s dalšími neurokutánními syndromy a postihuje přibližně stejně obě pohlaví. Onemocnění vzniká poruchou neurogliální proliferace a jeho etiologie není doposud přesně známa. HME se může projevit těžkou kraniofaciální asymetrií, psychomotorickou retardací, lateralizací, např. hemianopií, hemiparézou aj, a především těžkými epileptickými záchvaty v raném dětství, které bývají refrakterní na konzervativní způsob léčby. Diagnostika je postavena na klinickém obraze a MRI vyšetření, které vyloučí jiné příčiny makrocefalie u novorozenců nebo jiné unilaterální hemisferální léze. Cílem léčby je ovlivnění charakteru a počtu záchvatů. U těžké formy HME, provázené refrakterní křečí, je kauzální léčbou hemisférektomie.
V následujícím článku bychom chtěli připomenout HME jako nepříliš časté neurochirurgické onemocnění a dnes již velmi sporadicky užívanou chirurgickou metodu – hemisférektomii. V článku uvádíme naše zkušenosti s chirurgickou léčbou 5 nemocných dětí s HME během 3 let.
Metodika
Na Klinice dětské chirurgie a traumatologie 3.LF UK, FTN Praha, bylo v letech 2001-2004 operováno 5 dětí, 4 chlapci a 1 dívka. Průměrný věk nemocných byl v době operace 28 měsíců, věkového rozmezí 6 měsíců až 5 let u chlapců a 2měsíční dívka). Indikaci chirurgického výkonu se opírala o přítomnost častých farmakologicky refrakterních epileptických záchvatů a MRI-průkaz hemimegalencefalie. Podrobný přehled klinických nálezů u jednotlivých dětských nemocných ukazuje tab. 1.

Celkem bylo provedeno 7 chirurgických výkonů – u 2 dětí jsme provedli chirurgickou revizi. Typy výkonů jsou uvedeny v tab. 2.

Výsledky
U všech dětských nemocných došlo po operaci ke snížení počtu záchvatů a ke změně charakteru záchvatů. Jeden nemocný je rok po operaci zcela bez záchvatů, u jednoho nemocného jsou nyní patrny jen myoklonické záškuby očních víček. Hemiparéza se u všech nemocných postupně upravila, u dvou nemocných s psychomotorickou retardací na úroveň novorozence nedošlo ke zlepšení psychomotorického vývoje ani po chirurgickém výkonu. 30denní chirurgická mortalita v souboru je nulová. Zaznamenali jsme jednu chirurgickou komplikaci, vytvoření likvorové pseudocysty. Stav jsme řešili plastikou tvrdé pleny pomocí autotransplantátu z okolního periostu.
Diskuse
Poruchy vývoje mozkové kůry u člověka tvoří různotvárnou skupinu vrozených vývojových vad (hamartomatóz), u nichž je společným jmenovatelem anomální chování kmenové mozkové buňky – neuroblastu. Mozkové hamartomatózy bývají členěny na poruchy proliferace, migrace nebo nitrokorové organizace neuroblastů, případně poruchy kombinované. HME, jako benigní hamartomatóza, spadá mezi poruchy migrační [5]. Projevuje se asymetrií, způsobenou excesivním růstem jedné hemisféry a ztluštěním mozkové kůry (pachygyrií). HME se liší od ostatních mozkových dysgenezí, protože výsledná extrémní asymetrie u HME neodpovídá žádnému z vývojových stadií lidského mozku [6]. Poprvé byla HME popsána v r. 1835 v Simsově práci o mozkových hypertrofiích a atrofiích s nálezy z 253 provedených sekcí [7]. U HME (též unilaterální megalencefalie) lze dále morfologicky rozlišit 3 formy [8] podle rozsahu postižení.
- izolovaná HME - vyskytuje se vzácně a obvykle je spojena s hemikorporální hypertrofií, kožním či systémovým postižením
- syndromová HME – častější forma, bývá doprovázena dalšími formami fakomatóz (např. neurofibromatózou, tuberózní sklerózou, syndromem epidermálních névů aj)
- totální HME. Forma s ipsilaterální asymetrií kmene či mozečku. Přítomna může být také hemikorporální hypertrofie stejnostranně nebo hemifaciální hypertrofie na protilehlé straně.
Tradiční členění mozkových hamartomatóz vychází z pozorování embryonální poruchy migrace neuroblastů, která nastává ve 3.-5. týdnu gestace [9,10]. Podobně jako u tuberózní sklerózy jsou jednotlivé buňky alterovány v růstu a diferenciaci současně s poruchou tkáňové architektury a heterotopickým umístěním buněk. Novější studie prokázaly, že jde o výsledek časnější, geneticky naprogramované poruchy vývoje určité buněčné linie ve vztahu k symetrii. Z tohoto hlediska ztrácí tradiční členění na hodnotě, protože HME může být považována i za poruchu proliferace. Předpokládá se vztah mezi epidermálním růstovým faktorem a nadměrnou proliferací. Někteří autoři považují hemimegalencefalii za neoplazii [11]. V posledním desetiletí, s rozvojem molekulární genetiky, byly rozpoznány tzv. „organizing“ geny, které odpovídají za pravolevou symetrii neuraxis a závislého mezodermu (např. obratlových těl). Mitogeny, které upravují poměr synapticky vázaných neuronů, se mohou dostat do interakce s těmito geny pro symetrii. Popsaná genová interakce může podpořit vysvětlení vzniku HME, u níž je zvětšená hemisféra extenzivně celulární. Přesný popis role genových interakcí u konkrétní patologie, jakou je například HME, není úplně znám. Přesto mohou nové poznatky, zejména z oboru genetiky, sloužit jako teoretický základ pro vysvětlení různých typů vrozených mozkových asymetrií [12]. Dnes se do příčinné souvislosti s chováním genu pro symetrii klade řada vrozených vývojových onemocnění (např. Sturgeův-Weberův syndrom, unilaterální familiární = u. f. pachygyrie, u. f. schizencefalie, f. porencefalie, u. f. nodulární periventrikulární heterotopie, Parryova-Rombergova progresivní hemifaciální atrofie, Lhermittův–Duclosův dysplastický gangliocytom mozečku a další). HME postihuje všechny etnické skupiny a obě pohlaví přibližně stejně, někteří autoři připisují mírnou predominanci u mužů. Četnost postižení levé a pravé hemisféry je rovněž přibližně stejná. HME je nutno odlišit od infarktů, infekcí a jiných získaných onemocnění, která mohou způsobit u fetu asymetrii mozkových hemisfér. Onemocnění není sekundární a není u něj prokázána patofyziologická souvislost s metabolickými nebo neurodegenerativními onemocněními. U nemocných s HME nebyly nalezeny žádné chromozomální aberace [8,13].

HME se klinicky manifestuje obvykle v raném dětství, velmi často již v novorozeneckém období a klinický obraz bývá u všech 3 forem onemocnění stejný. Zahrnuje farmakorezistentní epileptické záchvaty, těžkou psychomotorickou retardaci, a neurologickou lateralizaci, často kontralaterální hemiparézu, hemianopii aj.
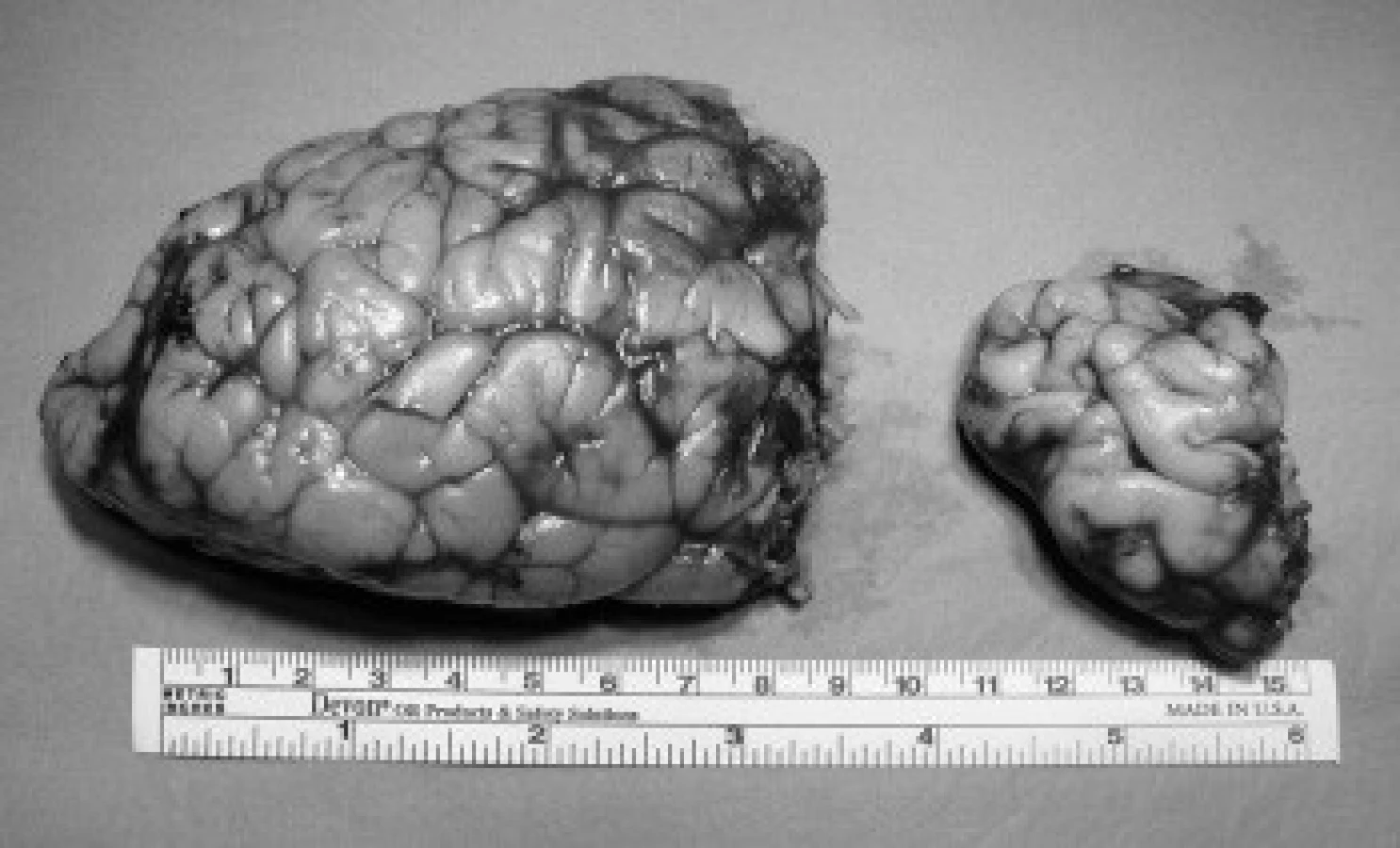
První zřetelnou klinickou známkou u izolované formy HME bývá makrocefalie. K abnormálnímu zvětšení obvodu hlavy dochází v časném údobí po porodu nebo v prvních měsících života. Tou dobou je u novorozence (případně kojence) s HME dobře patrna i asymetrie krania. Obličej postrádá dysmorfické rysy [8,10]. Náhlé enormní zvětšení neurokrania během prvních měsíců života může vést k diagnostickým omylům. Často bývá zvažován tumor, dále jsou známy případy mylné implantace VP-zkratu nemocným dětem s HME pro „hydrocefalus“, který nebyl nikdy potvrzen [14]. U všech dětí s časnými projevy HME je patrná celková vývojová retardace. Bývají výrazně sníženy kognitivní, zejména jazykové dovednosti – nemocní zvládnou obvykle jen několik slov či jednoduchých vět, popsána byla i úplná ztráta řečových funkcí [15]. Neurologická lateralizace v klinickém obraze HME může dosahovat značné míry, stupeň kontralaterální hemiparézy obvykle odpovídá tíží celkového postižení, extrémním vyjádřením je obraz generalizované hypotonie. Hemianopie je nalézána ipsilaterálně k postižené hemisféře. Nejčastějším projevem onemocnění je epilepsie, různé epileptogenní projevy nalezneme až u 93 % postižených. Projevy epilepsie se objevují již v prvních dnech života a obvykle jsou farmakorezistentní. Zahrnují různé typy záchvatů, od tonických křečí - tzv. Ohtaharův syndrom [16], přes parciální motorické nebo komplexní, někdy se sekundární generalizací. Infantilní spazmy se objevují u 50 % nemocných, myoklonie se objevují zřídka. Záchvaty někdy přecházejí do epileptického statu, který může v extrémních případech vést k úmrtí pacienta [17]. Syndromová HME se od izolované formy liší tím, že je vždy spojena se systémovým postižením, obvykle kutánní lézí. Popsána je celá řada neurokutánních syndromů, které se vzácně mohou vyskytnout s hemimegalencefalií: např. neurofibromatóza, tuberózní skleróza, syndrom epidermálních névů, protomelanóza, Klippelův-Trénaunayův-Weberův syndrom, Aicardiův syndrom, Itova hypomelanóza aj. Ojediněle byla popsána u Hirschprungovy choroby [14]. V našem souboru se jako iniciální příznaky manifestovaly epileptické záchvaty u všech nemocných dětí, u všech se jednalo o farmakorezistentní formu EP onemocnění. U 4 nemocných se onemocnění projevilo tonicko-klonickými křečemi se sekundární generalizací (tab. 1). U 1 nemocné došlo k rozvoji epileptického statu, který by řešen uvedením nemocné do řízeného thiopentalového komatu (nem. č. 3, tab. 1). U dalšího nemocného se onemocnění manifestovalo myoklonickými záškuby (nem. č. 5, tab. 1).
Diagnózu HME je třeba zvažovat již od prvních popsaných klinických příznaků. Diagnostickou metodou volby je MRI. Vyšetření potvrdí přítomnost, typ a stupeň HME, vyloučí jiné unilaterální léze, jako je krvácení, kongenitální tumor, cysty [14,18]. K typickým znakům HME na MRI-snímku patří asymetrie hemisfér a komorového systému, rozšíření jedné hemisféry a dysplazie (nejčastěji ztluštění) mozkové kůry. Postranní komory bývají deviovány (přesun střední čáry na protilehlou stranu v celém průběhu nebo jen okcipitálně, frontální roh protažen, okcipitální roh a trigonum dilatovány aj). U nemocných s HME nalezáme na MRI známky poruchy diferenciace šedé a bílé hmoty, korové dysplazie typu lissencefalie, pachygyrie, polymikrogyrie, schizencefalie či asymetrie kalózního tělesa. Různé typy popsaných patologických nálezů se mohou vyskytovat ojediněle nebo společně u 1 nemocného [18-20]. Podle stupně postižení na MRI-nálezu lze HME dělit na 3 stupně, členění respektuje i tíži klinického nálezu, proto se arbitrážně stanovené znaky pro jednotlivé stupně mohou překrývat.
I. stupeň: lehké rozšíření postižené hemisféry, lehká komorová asymetrie s protažením frontálního rohu postranní komory. Střední čára bez přesunu, hyperintenzity v bílé hmotě, kortikální dysplazie není zjevná.
II. stupeň: všechny znaky I. stupně jsou vyjádřeny větší měrou, zřetelná kortikální dysplazie
III. stupeň: výrazně zvětšená hemisféra s přesunem střední čáry, rozšíření postranní komory, rozsáhlá kortikální dysplazie.
V patologickém nálezu HME převažuje pachygyrie s okrsky polymikrogyrie. Šedá hmota korová může být až 3násobně ztluštělá, vrstva subkortikální bílé hmoty je rovněž zesílena, diferenciace mezi šedou a bílou hmotou je setřelá. Postranní komora bývá mírně dilatována, kalózní těleso ztenčeno. Heterotopické okrsky šedé hmoty lze nalézt v centrum semiovale a v temporálním laloku. Zadní mozek obvykle nebývá asymetrií postižen, někdy je patrna asymetrie pedunklů . Mikroskopicky jsou přítomny abnormality ve velikosti a cytomorfologii buněk bílé i šedé hmoty, buněčná architektura bývá porušena. Architektura kortexu je spíše sloupcová než ve vrstvách. Mnoho buněk, zejména ve spodních vrstvách kortexu, je zvětšeno s množstvím cytoplazmatických Nisslových tělísek. Postižené buňky nabývají bizarních tvarů, lze pozorovat balonovité neurony (podobně jako u Tailorovy kortikální dysplazie). Buňky v mitóze nebývají zastiženy. S-100 β protein je přítomen ve zvýšeném množství [15-20]. U všech 5 nemocných dětí našeho souboru se v grafickém nálezu (MRI) zobrazily typické znaky pro HME, tj. asymetrie hemisfér, asymetrie komorového systému, pachygyrie. Jiné typy korové dysplazie se v MRI-obraze našich nemocných nezobrazily (tab.1). Grafický MRI-nález odpovídal HME II. stupně u 4 nemocných, u 7měsíční dívky odpovídal nález HME III. stupně (nem. č. 2, tab. 1).
Terapeutickým cílem je ovlivnění počtu a charakteru záchvatů. Konzervativní terapie u lehkého a středně těžkého stupně postižení znamená kombinaci několika antiepiletik spolu s pečlivým sledováním sérových hladin léčiva. Chirurgická léčba je indikována u nemocných, u nichž selhává léčba farmakologická nebo u nemocných, u nichž dochází k výrazné kumulaci negativních účinků zvolené kombinace antiepileptik. K užívaným typům chirurgických zákroků patří:
- lobární multilobární resekce
- subpiální transsekce v motorické krajině
- dekortikace
- hemisférektomie
Kauzální léčbou těžké formy HME s refrakterními epileptickými záchvaty je hemisférektomie (HSE). HSE byla poprvé užita u nemocného s HME v roce 1978 [21]. Relativně invazivní resekční výkon –funkční HSE (přerušení funkčních spojů mezi hemisférami), případně anatomická HSE (odstranění postižené hemisféry s ponecháním basálních ganglií a zrakového kortexu), jsou metodou volby i přes uváděnou značnou morbiditu a mortalitu výkonu (různé práce uvádějí morbiditu 35-40% a mortalitu 2-4%). Indikační volba mezi anatomickou a funkční HSE je dosud předmětem diskuze [22-24]. V úvahu je brán nejen stupeň postižení, ale i věk a hmotnost nemocného dítěte. U nemocných s HME dává většina autorů přednost radikálnější anatomické HSE, tj. úplnému odstranění postižené hemisféry, i přes vyšší M/M výkonu. V případech HME s úbytkem mozkové tkáně volí řada autorů funkční HSE, tj. postižená hemisféra je ponechána in situ, chirurg přeruší pouze funkční spoje hemisféry [15,16]. Zmenšení počtu a změna charakteru záchvatů po výkonu přináší významné zlepšení kvality života pro nemocné i jejich okolí. Po úspěšném výkonu lze přiměřeně redukovat antiepileptickou medikaci, nezanedbatelný není tedy ani ekonomický přínos HME. Všichni nemocní v našem souboru byli před indikací k chirurgickému výkonu léčeni konzervativně, vždy s nulovým efektem. U všech nemocných souboru byla provedena chirurgické léčba, jednotlivé užité metody uvádí tab. 2. Výkon s největší mírou invazivity, anatomická HSE, byl proveden u 2 nemocných souboru. U 2 nemocných byla chirurgická léčba opakována, u 1 byl zvětšen rozsah předchozího zákroku, aniž byl změněn charakter zákroku (nem. č. 3, tab. 2), u 2. nemocného byla původně provedená funkční HSE ve druhé době rozšířena o anatomickou HSE (nem. č. 4, tab. 2). U nemocných po rozsáhlých epileptochirurgických výkonech závisí další psychomotorický rozvoj na stupni dosaženého PM vývoje před výkonem.V případech, v nichž se vývoj zastavil v novorozeneckém věku, chirurgická léčba další PM vývoj neovlivní. U nemocných s HME s nepřerušeným PM vývojem chirurgická léčba PM vývoj může zpomalit, ale obvykle jej nezastaví. V našem souboru u2 nemocných s pozastaveným PM vývojem na úrovni novorozence nedošlo k žádnému zlepšení ani při dlouhodobém sledování po výkonu (nem. č. 2 a 4, tab. 2). Komplikace spojené s výskytem hemosiderózy nebyly u nemocných našeho souboru zatím pozorovány.
Závěr
- Hemimegalencefalie je asymetrizující porucha vývoje CNS, může se morfologicky projevovat nejméně ve 3 odlišných formách, a to buď izolovaně, nebo sdružená s dalším, většinou neurokutánním systémovým postižením.
- Onemocnění se klinicky projevuje různými typy epileptických záchvatů s různou frekvencí a různou, někdy velmi výraznou intenzitou. Ve většině případů dosahuje četnost záchvatů až desítek týdně, u některých nemocných desítek denně. Záchvaty mohou vyústit ve status epilepticus, případně mohou vést až k úmrtí nemocného.
- Epileptogenní projevy HME je nutno řešit radikálně, účinnost konzervativní léčby je omezena, někdy nulová. Neúčinnost konzervativní léčby nutí ošetřujícího lékaře podávat kombinace antiepileptik, která mívají i negativní vedlejší účinky. Při neúčinnosti konzervativní léčby je metodou volby chirurgické řešení. Indikovány jsou i relativně invazivní metody.
- Přestože chirurgická léčba HME nemůže ovlivnit podstatu vrozené poruchy, má výrazný účinek na zlepšení kvality života postiženého dítěte a rovněž pro okolí nemocného. Zanedbatelným není ani ekonomický efekt léčby.
- Výsledky chirurgické léčby 5 nemocných dětí v našem souboru potvrzují účinnost metody.
Přijato k recenzi: 20. 6. 2006
Přijato do tisku: 5. 4. 2007
MUDr. Helena Homolková
Klinika dětské chirurgie a traumatologie 3. LF UK
Fakultní Thomayerova nemocnice,
140 59, Vídeňská 800, Praha 4
e-mail: hhomolkova@seznam.cz
Sources
1. Walsh CA. Genetics of neuronal migration in the cerebral cortex. MRDD Res Rev 2000; 6: 34–40.
2. Marin O, Rubenstein JLR. A long remarkable journey: tangential migration in the telencephalon. Nature Rev 2001; 2: 780–790.
3. Xu Q, Cobos I, De La Cruz E, Rubenstein JLR, Anderson SA. Origins of cortical interneuron subtypes. J Neurosci 2004; 24: 2612–2622.
4. Barkovich AJ, Raybaud CA. Neuroimaging in disorders of cortical development. Neuroimaging Clin N Am 2004; 14(2): 231–254.
5. Barkovich AJ, Kuzniecky RI, Jackson GD, Guerrini R, Dobyns WB. Classification system for malformations of cortical development: Update 2001. Neurology 2001; 57: 2168–2178.
6. Sarnat HB. Cerebral dysgenesis. New York: Oxford University Press 1992.
7. Sims J. On hypertrophy and atrophy of the brain. Med Quir Trans 1835; 19:315-318.
8. Trounce JQ, Rutter N, Mellor DH. Hemimegalencephaly: Diagnosis and treatment. Dev Med Child Neurol 1991; 33: 261-266.
9. Takashima S, Chan F, Becker LE, Kuruta H. Aberrant neuronal development in hemimegalencephaly: Imunohistochemical and Golgi studies. Pediatr Neurol 1991; 7: 275-280.
10. Woo CL, Chuang SH, Becker LE, Jay V, Otrubo H, Rutka ST et al. Radiologic- pathologic correlation in focal cortical dysplazia and hemimegalencephaly in 18 children. Pediatr Neurol 2001; 25: 259-303.
11. Townsend JJ, Nielsen SL, Malamud N. Unilateral megalencephaly : Hamartoma or neoplasm? Neurology 1975; 25: 448-453.
12. Saijoh Y, Adachi H, Mochida K, Ohishi S, Hirao A, Hamada H et al. Distinct transcriptional regulatory mechanism underlie left-right asymetric expression of lefty 1 and lefty 2. Genes Dev 1999; 13: 259-269.
13. Battaglia D, Di Rocco C, Iuvone L, Acquafondata C, Ianelli A, Lettori D et al. Neuro- cognitive development and epilepsy in children with surgically treated hemimegalencephaly. Neuropediatrics 1999; 30: 307-313.
14. Thong MK, Thompson E, Keenan R, Sommer K, Harbord M, Davidson G et al. A child with hemimegalencephaly, hemihypertrophy, macrocephaly, cutaneous vascular malformation, psychomotor retardation and intestinal lymphangiectasia – A diagnostic dilemma. Clin Dysmorphol 1999; 8: 283-286.
15. Flores-Sarnat L. Hemi-megalencephaly (Part 1): Genetic, clinical and imaging aspects. J Child Neuro 2002; 17: 373-374.
16. Ohtsuka Y, Ohno S, Oka E. Electroclinical characteristics of hemimegalencephaly. Pediatr Neurol 1999; 20: 390-393.
17. Reardon W, Harding B, Winter B, Baraitzer M. Hemihypertrophy, hemimegalencephaly, and polydactyly. Am J Med Genet 1996; 66: 144-146.
18. Cho W, Seidenwurm D, Barkovich AJ. Adult onset of neurologic dysfunctionassociated with cortical malformations. AJNR Am J Neuroradiol 1999; 20: 1037–1043.
19. Flores-Sarnat L, Sarnat HB, Gutierez GD, Alvarez A. Hemimegalencephaly: Part 2.Neuropathology Suggest a Disorder of Cellular Lineage . J Child Neurol 2003; 18: 776-785.
20. Yasha TC, Santosh V, Das S, Shankar SK. Hemimegalencephaly: Morphological and immunohistochemical study. Clin Neuropathol 1997; 16: 17-22.
21. Fitz CR, Harwood-Nash DC, Boldt DW. The radiographic features of unilateral megalencephaly. Neuroradiology 1978; 15: 145-148.
22. Di Rocco C, Iannelli A. Hemimegalencephaly and intractable epilepsy: complication of hemispherectomy and their correlations with surgical technique. A report on 15 cases. Pediatr Neurosurg 2000; 33(4): 198-207.
23. Zentner J. Surgical treatment of epilepsies. Acta Neurochir Suppl 2002; 84: 27-35.
24. Carreno M, Wyllie E, Bingaman W, Kotagal P, Comair Y, Ruggieri P. Seizure outcome after functional hemispherectomy for malformations of cortical development. Neurology 2001; 57(2): 331-333.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2007 Issue 5
Most read in this issue
- Léčba epileptických syndromů u dětí
- Hodnocení edému terče zrakového nervu
- Jsou některé kontraindikace lumbální punkce dnes již obsoletní? Kazuistika
- Transforaminální lumbo-sakrální mezitělová fúze (TLIF) s instrumentací: prospektivní studie s minimálně 20měsíčním sledováním