
Diferenciální diagnostika neuromyelitis optica a onemocnění jejího širšího spektra
Differential diagnosis of neuromyelitis optica spectrum disorders
Differential diagnosis of autoimmune CNS disorders is facilitated by the assessment of aquaporin-4 antibodies (AQP4-IgG) and myelin oligodendrocyte glycoprotein (MOG-IgG). These autoantibodies are associated with neuromyelitis optica spectrum disorders (NMOSD) and MOG encephalomyelitis, respectively. Furthermore, these antibodies can confirm a considered diagnosis. The diagnosis of NMOSD can be based on clinical manifestation, e. g., severe optic neuritis and/or complete transverse myelitis. Nevertheless, the differential diagnosis of NMOSD is often challenging in incomplete transverse myelitis, brainstem symptoms, and optic neuritis with a good response to corticosteroid treatment. MRI findings and the absence of oligoclonal bands in cerebrospinal fluid can lead to a correct diagnosis. The most common disorders in the differential diagnosis for NMOSD are MS, ischemic lesions, and tumors.
Keywords:
MOG encephalomyelitis – neuromyelitis optica spectrum disorders
Autoři:
P. Nytrová
Působiště autorů:
Neurologická klinika a Centrum klinických neurověd, 1. LF UK a VFN v Praze
Vyšlo v časopise:
Cesk Slov Neurol N 2020; 83/116(supplementum 1): 51-57
doi:
https://doi.org/10.14735/amcsnn2020S51
Souhrn
Diferenciální diagnostika autoimunitních onemocnění CNS se zjednodušuje díky stanovení protilátek proti akvaporinu-4 (AQP4-IgG) nebo myelinovému oligodendrocytárnímu glykoproteinu (MOG-IgG). Tyto protilátky jsou asociovány s neuromyelitis optica a onemocněním jejího širšího spektra (neuromyelitis optica spectrum disorders; NMOSD), respektive MOG encefalomyelitidou, a mohou potvrdit zvažovanou diagnózu. Často uvažujeme o diagnóze NMOSD na základě klinického obrazu, jako např. u těžkých optických neuritid a/nebo transverzálních míšních lézí. Složitější může být diferenciální diagnostika u parciálních míšních syndromů, kmenové symptomatiky nebo optické neuritidy s rychlou odpovědí na terapii kortikosteroidy. Tam nás mohou ke správné diagnóze spíše navést nálezy na MR a případně absence oligoklonálních pásů v likvoru. V diferenciální diagnostice nejčastěji zvažujeme RS, vaskulární léze a neoplazie.
Klíčová slova:
neuromyelitis optica a onemocnění jejího širšího spektra – MOG encefalomyelitida
Úvod
Z pohledu diferenciální diagnostiky je vhodné se na neuromyelitis optica (NMO) podívat jako na syndrom, protože dnes již víme, že nemusí být zprostředkován pouze protilátkami proti akvaporinu-4 (AQP4-IgG), ale může být asociován i s protilátkami proti myelinovému oligodendrocytárnímu glykoproteinu (MOG-IgG), případně se protilátky neprokážou [1,2]. I přes podobný klinický obraz jsou u těchto skupin odlišnosti v patogenezi, terapeutické odpovědi i prognóze pacientů. Původní koncept syndromu NMO zahrnoval pouze optickou neuritidu (ON) a akutní myelitidu. Dnes je spektrum klinických příznaků širší a akceptují se příznaky způsobené zánětlivým postižením i mimo optické nervy a míchu. Neuromyelitis optica a onemocnění jejího širšího spektra (neuromyelitis optica spectrum disorders; NMOSD) pak zahrnuje i různé kmenové příznaky (např. syndrom area postrema) nebo méně obvykle diencefalický či cerebrální syndrom [3].
Diagnostiku můžeme rozdělit do dvou kroků. První krok zahrnuje určení základní etiologie klinického syndromu ve smyslu vaskulární, infekční, autoimunitní, neoplastické, postradiační, metabolické a další. V druhém kroku v rámci autoimunitní etiologie postižení CNS určujeme diagnózu, která může být součástí orgánově nespecifických systémových autoimunit s postižením CNS (např. systémový lupus erythematodes [SLE], sarkoidóza atd.) nebo orgánově specifických onemocnění (RS, akutní diseminovaná encefalomyelitida [ADEM], NMOSD, MOG encefalomyelitida [MOG-EM] a další).
Nezastupitelné místo v diagnostickém procesu pak mají vyšetření MR a laboratorní vyšetření, kde dominuje stanovení autoprotilátek, sérologické a PCR testy a případně vyšetření mozkomíšního moku. Mezi další vyšetřovací modality užívané v diagnostice patří evokované potenciály a optická koherenční tomografie.
V klinickém obraze může podporovat diagnózu autoimunitního postižení CNS koincidence s jiným autoimunitním onemocněním, a to zejména ze skupiny revmatologických onemocnění a idiopatických střevních zánětů.
Určení správné nozologické jednotky a poté terapie je zcela zásadní pro odvrácení negativní prognózy těchto pacientů. Musíme pečlivě hodnotit celkový stav pacienta, předchorobí i rodinnou anamnézu.
Diferenciální diagnostika myelitid
Typicky se u pacientů s NMOSD rozvíjí myelitida během dnů, někteří pacienti udávají nespecifické prodromální příznaky jako únavu, pocení, někdy horečky, bolest v krku či nespecifická virová onemocnění. U jiných pacientů v anamnéze nacházíme operační výkon (pravděpodobně dochází k ovlivnění propustnosti hematoencefalické bariéry anestetiky), někdy se první příznaky nemoci mohou objevit v graviditě nebo poporodním období. Přehled diferenciální diagnostiky akutních myelitid shrnuje tab. 1.
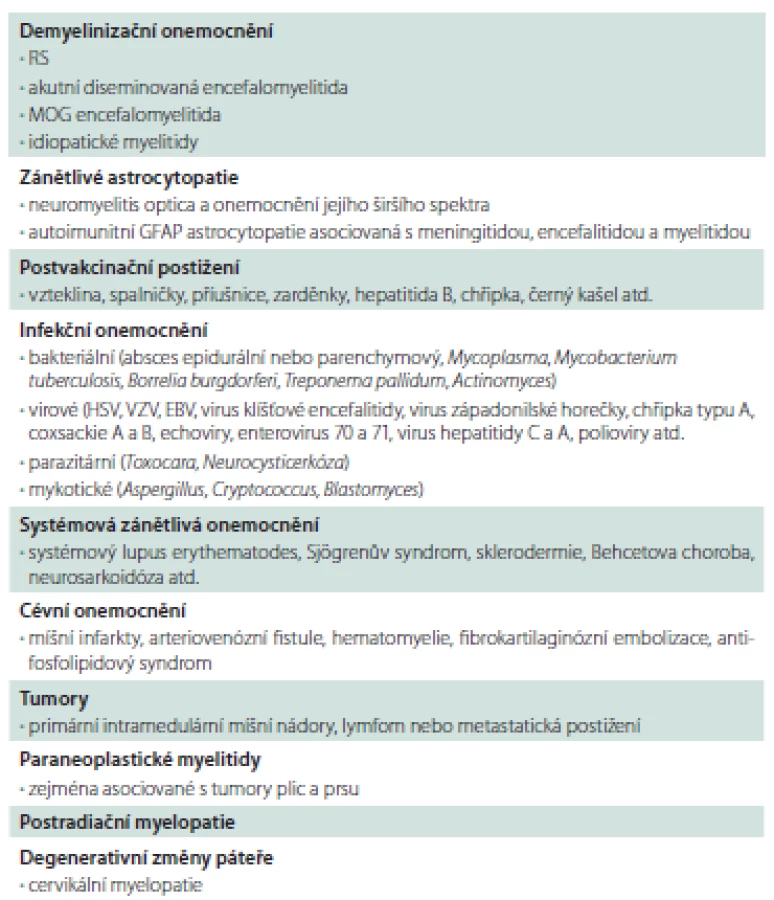
Klinicky se u myelitidy setkáváme s obrazem transverzální míšní léze (TML) i parciálních míšních syndromů. U akutně vzniklé TML netraumatické etiologie je vždy indikováno vyšetření AQP4-IgG a MOG-IgG v séru. Myelitida pod obrazem TML je typickým prvním příznakem NMOSD u pacientů starších 50 let [4]. U mladších pacientů se spíše iniciálně setkáváme s ON. Na T2 vážených obrazech na MR páteře vidíme dlouhá ložiska dosahující nebo přesahující 3 obratlové segmenty, tzv. „longitudinally extensive transverse myelitis“ (LETM). Ta mívá u NMOSD pacientů vřetenovitý tvar a může zasahovat až do oblasti prodloužené míchy. V akutních stadiích často vidíme sycení kontrastní látkou [3]. Nutno dodat, že ložiska zvýšeného signálu na T2 vážených obrazech přesahují i délku šesti obratlových těl. Na transverzálních řezech vidíme výrazné postižení šedé hmoty míšní [5]. LETM s pozitivitou MOG-IgG bývá častěji lokalizována v oblasti míšního konu a epikonu s klinicky dominujícím postižením sfinkterů v porovnání s AQP4-IgG pozitivní LETM [6]. Po úspěšné terapii dochází k výrazné regresi nálezu, což napomáhá v diferenciální diagnostice u míšních tumorů kdy nález neregreduje a není přítomna výrazná atrofie míchy. V mozkomíšním moku se u pacientů s tumorem můžeme setkat s přítomností erytrocytů v cytologickém nálezu nebo s různými rozpadovými produkty hemoglobinu při spektrofotometrickém vyšetření, což v klinickém obraze bývá doprovázeno rozvojem bolestí zad a pozitivními meningeálními jevy. To je dáno přítomností drobných subarachnoidálních krvácení u míšních tumorů [7]. Toto je potřeba odlišit od bolestivých spasticko-dystonických paroxyzmálních tonických křečí trupu i končetin u pacientů s myelitidou a pozitivitou AQP4-IgG, které se objevují přibližně 4–6 týdnů od prvních příznaků. Křeče dobře reagují na terapii karbamazepinem [8]. Tyto paroxysmální tonické stavy většinou trvají desítky vteřin a objevují se během dne s různou intenzitou, častěji v závislosti na pohybu. U primárních intramedulárních tumorů dochází k subakutní až chronické progresi obtíží. Klinické obtíže mohou začínat syringomyelitickou disociací čití. U NMOSD je jasný atakovitý průběh. U transverzální myelitidy s pozitivitou AQP4-IgG nacházíme v mozkomíšním moku typicky cytoproteinovou asociaci při poruše hematoencefalické bariéry během ataky [4]. Nález se normalizuje v remisi.
Obraz akutní myelitidy mohou způsobit různé patogeny, přičemž zde mezi nejčastější infekční agens řadíme především herpes-viry, enteroviry a další. Akutní myelitida provázená obrazem chabé parézy s nálezem lézí v šedé hmotě míšní („polio-like“ syndrom) je nyní popisována v souvislosti s infekcemi enterovirem 68, enterovirem 71 a coxsackie virem A7 [9]. V diagnostice infekční etiologie myelitidy se neobejdeme bez sérologických a PCR testů, případně kultivace mozkomíšního moku. I zde na MR může být obraz LETM. Je dobré si uvědomit, že ne všechny LETM nutně znamenají diagnózu NMOSD. Naopak ani myelitidy s ložiskem nedosahujícím tří obratlových segmentů při zobrazení pomocí MR diagnózu NMOSD nevylučují.
Vzácněji LETM nalezneme u pacientů s RS, ale zde v naprosté většině vznikají splýváním jednotlivých kratších lézí v čase, a proto je potřeba hodnotit i jednotlivé MR snímky z dřívějších vyšetření. U RS častěji vidíme v klinickém obraze v rámci akutní myelitidy některý z parciálních míšních syndromů a asymetrické postižení. Také u pacientů s NMOSD se můžeme setkat s tímto klinickým obrazem. Určení správné diagnózy je pak komplikovanější, zejména když se jedná o iniciální příznak onemocnění. V těchto případech netestujeme u všech jedinců AQP4-IgG nebo MOG-IgG. Je potřeba se podívat na nálezy v mozkomíšním moku, kde je u RS většinou oligocytóza a přítomnost oligoklonálních pásů (oligoclonal bands; OCB) pouze v likvoru. Koncentrace bílkoviny bývá normální nebo hraničně zvýšená. Absence OCB je typická pro NMOSD nebo MOG-EM. Počty elementů jsou vyšší, nejčastěji v desítkách buněk v jednom µl, typická je i přítomnost granulocytů. Nález OCB v mozkomíšním moku diagnózu NMOSD nevylučuje. Setkali jsme se s případy, kdy OCB v kontrolní lumbální punkci s odstupem času v řádu měsíců vymizely. Při atace bývá vyšší koncentrace bílkoviny a porucha hematoencefalické bariéry [4,10]. V těchto případech i u krátkých míšních lézí je vyšetření autoprotilátek plně indikováno. Na MR míchy u RS vidíme jinou distribuci ložisek na axiálních řezech, převážně v bílé hmotě, což je v kontrastu s NMOSD, kde dominuje postižení šedé hmoty míšní [5]. V takovém případě je nezbytné doplnění MR mozku. Typická ložiska pro RS jsou dobře známá. Nicméně se uvádí, že 8–10 % pacientů s NMOSD splňuje kritéria MR pro RS. U NMOSD nenacházíme typické léze periventrikulárně a v corpus callosum (CC), které by měly ovoidní tvar s osou kolmou na komory a v CC vytvářely tzv. Dawsonovy prsty. Léze v této lokalitě mohou být, ale mají zcela jiný charakter. Periventrikulární ložiska kopírují tvar komor. U NMOSD bývají nejčastěji supratentoriálně ložiska asymptomatická. Nicméně u relativně vzácných cerebrálních syndromů vidíme na MR mozku rozsáhlá ložiska v obou hemisférách, a to ne zcela jasně ohraničená. Tyto léze mají při úspěšné terapii tendenci k výrazné regresi [11].
Akutní myelitidy se také vyskytují u systémových zánětlivých onemocnění. Příkladem může být např. lupusová myelitida. Ta může být i prvním příznakem SLE a je zde potřeba pátrat po dalších příznacích nemoci (fotosenzitivita, kožní změny, postižení ledvin a další příznaky) a doplnit vyšetření autoprotilátek (proti nukleárním antigenům, dvouvláknové DNA, nukleosomům a dalším) [12]. Asi polovina pacientů s myelitidou a SLE má pozitivitu AQP4-IgG a jedná se o koincidenci dvou onemocnění (NMOSD a SLE) [13]. Myelitida může být i komplikací vaskulitid (primárních i sekundárních), ale ve většině případů je současně postižen i mozek [14].
Obtížnější bývá diagnostika ischemických lézí míchy. Zejména tam, kde není např. anamnéza kardiochirurgické operace, cévní rizika apod. Významným faktem je časový rozvoj obtíží – často se celý klinický obraz vyvíjí velmi dynamicky. V úvodu se objevuje i prudká bolest za krkem nebo v zádech vyzařující radikulárně. V dalších minutách až hodinách se rozvíjí porucha motoriky a čití i autonomní dysfunkce (sfinkterové a další) [15]. Zpočátku bývá obraz pseudochabé para/kvadruparézy či plegie – stadium míšního šoku. U některých pacientů můžeme v anamnéze vypátrat míšní klaudikace [16]. Klinicky nejčastěji vidíme ischemii přední spinální tepny, následovanou lézí předních rohů míšních, syndromem zadní míšní tepny a další [17]. Příčiny mohou být různé, mezi nejčastější řadíme disekce aorty nebo operační výkony na této tepně, případně jiné kardiochirurgické operace (s prolongovanou hypotenzí) a operace skolióz. Vzácně se popisují myelopatie u surfařů začátečníků a raritně u dospívajících gymnastek provádějících salta vzad [18,19]. Možné „vaskulární“ trauma (bez přítomnosti jasných traumatických změn okolních struktur) je popisováno i u jiných sportů. Mechanizmem je vysoké axiální zatížení nebo velká tlaková síla působící na temeno hlavy zejména při mírné flexi krku [20]. Na MR míchy v prvních hodinách od počátku obtíží nejsou často žádné změny. Při dalším vyšetření MR v odstupu 48–72 h je nález již plně vyjádřen s typickými znaky na T2 vážených obrazech, např. „soví oči“ při ischemii v povodí přední míšní arterie na axiálních řezech [15]. Zobrazení difúze pomocí MR (diffusion weight-ed imaging; DWI) může také napomoci k diagnostice míšní ischemie, kdy dochází k poklesu ADC (apparent diffusion coefficient) hodnoty v místě léze oproti nepostiženému úseku [21]. Nález v likvoru v prvních hodinách obtíží bývá často normální. I vaskulární léze mohou být autoimunitní etiologie, jedná se zejména o pacienty s antifosfolipidovým syndromem, u kterých tyto protilátky přímo interagují s endotelem cév a funkcí destiček. Stěžejní je průkaz těchto protilátek a také i cévního uzávěru (arteriálního nebo venózního), popř. nacházíme u žen v gynekologické anamnéze potraty po 10. týdnu těhotenství a další porodní komplikace [22]. Na kůži pacientů můžeme nalézt změny charakteru liveda reticularis, laboratorně může být přítomna v akutní fázi trombocytopenie.
Paraneoplastické myelitidy mohou mít akutní i chronický průběh. V druhém případě je diferenciálně diagnosticky zvažována primárně progresivní RS [23].
Myelopatie s dominující ataxií při postižení zadních provazců a případně spastickou paraparézou může být kromě deficitu vitamínu B12 dána i nedostatkem mědi. I u těchto případů spíše převažuje chronický průběh, ale může dojít i k akutní manifestaci. Měli bychom doplnit skrínink na celiakii či odhalit jiné příčiny těchto deficitů [24,25].
Diferenciální diagnostikaoptické neuritidy
Optická neuritida je provázená poruchou vizu rozvíjející se během hodin nebo několika dnů. Maximum obtíží se většinou manifestuje do 7–10 dnů. Tíže poruchy kolísá od rozmazaného vidění přes ztrátu centrálního vizu až k amauróze. Typické jsou také různé výpadky perimetru. Mezi další typické příznaky patří zhoršení barvocitu a kontrastní senzitivity. Pacient si stěžuje na bolest v oblasti oka nebo retroorbitálně, která se akcentuje pohybem bulbu. Při klinickém vyšetření bývá na postiženém oku přítomen relativní aferentní pupilární defekt (RAPD) [26]. U dvou třetin pacientů je normální nález na fundu. U třetiny pacientů bývá přítomen otok papily. Ve fázi rekonvalescence se může u některých pacientů objevovat např. Uhthoffův fenomén, kdy dochází k přechodnému zhoršení vizu v důsledku tepla nebo při zahřátí během cvičení. Méně známý je Pulfrichův fenomén, kdy dochází ke zhoršení prostorového vidění pohybujících se objektů [27]. V diferenciální diagnostice je potřeba zvážit, zda se jedná o ON nebo o některou z jiných diagnóz či autoimunitně podmíněných neuropatií zrakového nervu (tab. 2).
![Diferenciální diagnostika postižení zrakového nervu. Volně dle Weerasinghehoa a Luecka [26].](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image_pdf/99b99a4b7134cabfc4307fe408726ad0.png)
![Tab. 2 – pokračování. Diferenciální diagnostika postižení zrakového nervu. Volně dle Weerasinghehoa a Luecka [26].](https://pl-master.mdcdn.cz/media/image_pdf/a40b3b1b245c643982966bb526444e24.png?version=1606317992)
Optická neuritida může být příznakem RS, NMOSD nebo MOG-EM. Jejich základní charakteristiky jsou uvedeny v tabulce (tab. 3). Někdy se setkáváme s pacienty s idiopatickými recidivujícími ON, kde další bližší zařazení k nozologickým jednotkám není možné.
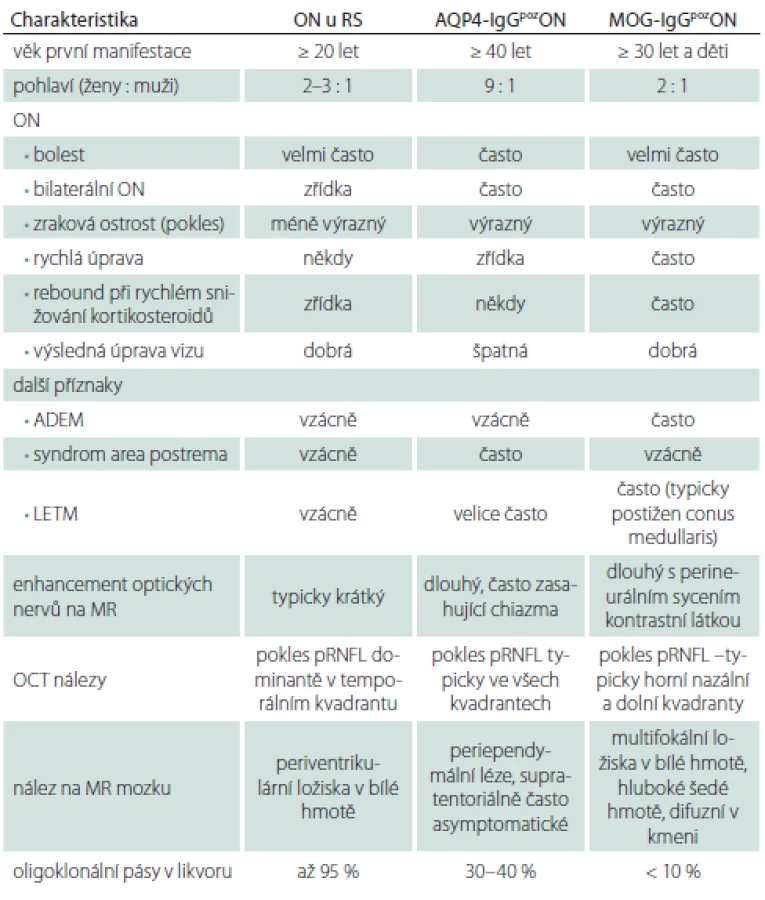
Nejčastěji se unilaterální ON vyskytuje u pacientů s RS nebo může jít o první příznaky této nemoci a hovoříme o klinicky izolovaném syndromu v případě, že vyloučíme jiné možné příčiny obtíží. Tito pacienti pak mají vysoké riziko přechodu do RS. Pro ně pak platí plně charakteristika ON uvedená výše. Často u těchto pacientů dochází k výrazné úpravě zrakových funkcí i bez terapie kortikosteroidy. Ty většinou urychlují návrat zrakových funkcí [28]. Pokud není úprava i přes terapii kortikosteroidy uspokojivá nebo dochází k další progresi obtíží, je vždy potřeba zvažovat jiné diagnózy. Je nezbytné provedení MR mozku a minimálně také krční míchy, kdy pátráme po hyperintenzitách na T2 vážených obrazech. To platí i pro situace bilaterálních ON. Ty jsou typické pro pacienty s pozitivitou MOG-IgG. Zde je i přes poměrně výrazné postižení zrakových funkcí velmi dobrá odpověď na terapii kortikosteroidy. Často je ON kortikodependentní, při rychlém snižování perorálních kortikosteroidů se však znovu objevuje vzplanutí onemocnění [26,28]. Pokud u pacienta s ON prokážeme v séru MOG-IgG a pomocí MR optických nervů nebo vizuálních evokovaných potenciálů potvrdíme zánětlivou lézi zrakového nervu, pak pacient naplní diagnostická kritéria pro MOG-EM [6]. Podobně u AQP4-IgGpozNMOSD vidíme bilaterální, nebo těsně sekvenčně za sebou následující ON. Zde však prakticky vždy i přes maximální terapii nedochází k úplné úpravě zrakových funkcí. Jsou popsány i případy, kdy se rozvine slepota po jediné atace této nemoci [29]. Tíže postižení zrakového nervu tak koreluje s poklesem peripapilární sítnicové vrstvy nervových vláken (pRNFL) měřené pomocí optické koherenční tomografie. Tento pokles je výrazný ve všech kvadrantech [4,30].
U bilaterálního postižení zraku je třeba myslet i na dědičná onemocnění, zejména na Leberovu hereditární atrofii optiku (LHON). Obtíže se rozvíjejí subakutně a většinou je postiženo nejprve jedno oko a po několika týdnech i druhé. Nebývají narušeny zornicové reflexy a většinou jde o nebolestivou postupně progredující poruchu vizu [26]. V diagnostice může napomoci fakt, že v akutní fázi bývá vstupní MR mozku normální. Nicméně stále není objasněna otázka možné asociace LHON s RS (tzv. syndrom Hardingové) [31]. Část pacientů s LHON však rozvíjí klinický i radiologický obraz neodlišitelný od RS (periventrikulární léze v bílé hmotě, přítomnost oligoklonálních pásů v likvoru) [32,33].
Je nutné si uvědomit, že rozvoj těžké ON u pacientů s pozitivitou AQP4-IgG může být rychlý, i v rámci hodin, a může imitovat cévní léze. První manifestace NMOSD může být v kterékoli dekádě života. U seniorů může dojít vzhledem k věku k diagnóze vaskulární léze. Také u ON s počátkem do měsíce po vakcinaci je potřeba stanovit zejména MOG-IgG, protože se může jednat o nespecifický inzult, který vedl k manifestaci autoimunitního onemocnění [6].
Oboustranné oční postižení vidíme i u chronické zánětlivé relabující neuropatie optiku (chronic relapsing inflammatory optic neuropathy; CRION). Zde je důležité vyloučení definovaného autoimunitního onemocnění, kde je tato manifestace jeho součástí, např. NMOSD (vč. negativity AQP4-IgG a MOG-IgG), RS a další nemoci. Pro tuto jednotku je typický návrat obtíží při snižování dávky nebo vysazení kortikosteroidů. Na MR je vidět sycení zrakového nervu kontrastní látkou [34].
Autoimunitní postižení zrakových nervů se mohou vyskytovat i u systémových onemocnění pojiva, jakými jsou např. SLE, Sjögrenův syndrom a další. Zde je situace jednodušší, pokud dochází k rozvoji zrakových obtíží u již známého onemocnění. Je třeba si uvědomit, že může jít i o komplikace léčby nebo infekční etiologii u výrazně imunosuprimovaných jedinců. U pacientů bývají vyjádřeny další celkové příznaky nemoci, jako jsou hubnutí, teploty, bolesti kloubů, sicca syndrom a další. Stejně jako myelitida ani ON nemusí být komplikací revmatologického onemocnění, ale může se jednat o koincidenci s NMOSD [35].
Někdy může dojít k postižení zrakového nervu granulomatózním procesem, který se může šířit z dutin, sinus cavernosus nebo granulomy vznikají v oblasti orbity (zejména apexu). Zde klinicky vidíme i postižení dalších hlavových nervů (obraz Tolosa-Hunt syndromu). Může se jednat o limitované formy ANCA vaskulitidy (bez postižení plic či ledvin) a diferenciálně diagnosticky zde zvažujeme jiné expanzivní procesy. Úzká spolupráce s ORL a očními specialisty je na místě, vč. provedení biopsie.
U infekčních zánětů je vždy potřeba provést adekvátní laboratorní vyšetření a úzce spolupracovat s oftalmology a infektology. Infekce mají často systémové projevy, po kterých musíme aktivně pátrat.
Diferenciální diagnostika kmenových, diencefalických a cerebrálních syndromů
Postižení mozkového kmene může být také iniciálním příznakem NMOSD.
Mezi dominující kmenové příznaky u APQ4-IgGpozNMOSD řadíme syndrom area postrema při postižení dorzální části prodloužené míchy v místě s vysokou hustotou exprese AQP4. Syndrom area postrema je charakterizován úpornou nauzeou, zvracením nebo singultem. Tyto obtíže mohou trvat i několik týdnů. Jako první manifestace NMOSD se objevují kmenové příznaky u 15,5–37 % pacientů [4,36]. Často jsou pacienti vyšetřování internisty s různým závěrem např. gastroenteritida, hiátová hernie, psychogenní etiologie a další. Syndrom area postrema se může vyskytnout kdykoli v průběhu onemocnění. Typické je, že použití symptomatické terapie selhává. Dobrá odpověď je na terapii kortikosteroidy nebo jinou imunoterapii. V roce 2015 byl syndrom area postrema zařazen mezi časté jádrové klinické charakteristiky NMOSD [37]. Pokud u tohoto syndromu prokážeme pozitivitu AQP4-IgG, pacient naplní kritéria pro NMOSD. Na MR na T2 vážených obrazech typicky nacházíme hyperintenzity v oblasti dorzální části medulla oblongata. U NMOSD se mohou vyskytovat i další kmenové příznaky – diplopie, vestibulární syndrom, dysartrie, paréza lícního nervu atd. Ty mohou být relativně častým příznakem i jiných onemocnění, např. RS. V těchto případech je zásadní zhodnocení charakteru ložiska na MR mozku. Pokud je ložisko v kontaktu se IV. komorou či periakveduktálně, pak bychom měli vždy stanovit AQP4-IgG v séru, a to i pokud nacházíme OCB v likvoru. U pacientů s kmenovou symptomatologií dochází nejčastěji ke stanovení chybné diagnózy RS. Nasazení chronické imunomodulační léčby určené pro pacienty s RS může u NMOSD vyvolat život ohrožující relaps.
U cerebrálních syndromů, projevujících se různou symptomatologií hemiparéz apod., je velmi těžké z klinického obrazu určit, zda by se mohlo jednat o NMOSD. Ložiska na MR jsou často neostře ohraničená, edematózní a přítomná v obou hemisférách. Napomoci nám může nález v mozkomíšním moku, kde nacházíme výraznou pleocytózu s přítomností granulocytů.
Narkolepsie jako příznak NMOSD je velmi vzácná, ale vždy je provázena nálezem lézí v oblasti diencefala, které jsou v kontaktu s III. mozkovou komorou.
Závěr
Diagnostika NMOSD se v posledních letech výrazně zlepšila. Nicméně stále existují situace, kdy je stanovení diagnózy výzvou. Pečlivost hodnocení klinických projevů spolu s nálezy na MR a v likvoru snižují riziko chybné diagnózy. Nejčastěji zvažované diagnózy pro podobný klinický obraz, nález na MR a v likvoru jsou RS, tumor nebo ischemické léze.
Grantová podpora
Podpořeno projektem Progres Q27/LF1.
Konflikt zájmů
Autoři deklarují, že v souvislosti s předmětem studie nemají žádný konflikt zájmů.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
MUDr. Petra Nytrová, Ph.D.
Neurologická klinika a Centrum klinických neurověd
1. LF UK a VFN v Praze
Kateřinská 30
128 08, Praha
e-mail: petra.nytrova@lf1.cuni.cz
Zdroje
1. Lennon VA, Kryzer TJ, Pittock SJ et al. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med 2005; 202 (4): 473–477. doi: 10.1084/jem.20050304.
2. Kitley J, Waters P, Woodhall M et al. Neuromyelitis optica spectrum disorders with aquaporin-4 and myelin-oligodendrocyte glycoprotein antibodies: a comparative study. JAMA Neurol 2014; 71 (3): 276–283. doi: 10.1001/jamaneurol.2013.5857.
3. Wingerchuk DM, Banwell B, Bennett JL et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015; 85 (2): 177–189. doi: 10.1212/WNL.0000000000001729.
4. Nytrová P, Kleinová P, Preiningerová Lízrová J et al. Neuromyelitis optica a poruchy jejího širšího spektra – retrospektivní analýza klinických a paraklinických nálezů. Cesk Slov Neurol N 2015; 78/111 (1): 72–77. doi: 10.14735/amcsnn201572
5. Nakamura M, Miyazawa I, Fujihara K et al. Preferential spinal central gray matter involvement in neuromyelitis optica: an MRI study. J Neurol 2008; 255: 163–170. doi: 10.1007/s00415-008-0545-z.
6. Jarius S, Paul F, Aktas O et al. MOG encephalomyelitis: international recommendations on diagnosis and antibody testing. J Neuroinflammation 2018; 15 (1): 134. doi: 10.1186/s12974-018-1144-2.
7. Dijindjian M, Djindjian R, Houdart R et al. Subarachnoid hemorrhage due to intraspinal tumors. Surg Neurol 1978; 9 (4): 223–229.
8. Carnero Contentti E, Leguizamón F, Hryb JP et al. Neuromyelitis optica: association with paroxysmal painful tonic spasms. Neuromielitis óptica: asociación con espasmos tónicos paroxísticos dolorosos. Neurologia 2016; 31 (8): 511–515. doi: 10.1016/j.nrl.2014.12.001.
9. Helfferich J, Knoester M, Van Leer-Buter CC et al. Acute flaccid myelitis and enterovirus D68: lessons from the past and present. Eur J Pediatr 2019; 178 (9): 1305–1315. doi: 10.1007/s00431-019-03435-3.
10. Jarius S, Paul F, Franciotta D et al. Cerebrospinal fluid findings in aquaporin-4 antibody positive neuromyelitis optica: results from 211 lumbar punctures. J Neurol Sci 2011; 306 (1–2): 82–90. doi: 10.1016/j.jns.2011.03.038.
11. Kim HJ, Paul F, Lana-Peixoto MA et al. MRI characteristics of neuromyelitis optica spectrum disorder: an international update. Neurology 2015; 84 (11): 1165–1173. doi: 10.1212/WNL.0000000000001367.
12. Pittock SJ, Lennon VA, de Seze J et al. Neuromyelitis optica and non organ-specific autoimmunity. Arch Neurol 2008; 65 (1): 78–83. doi: 10.1001/archneurol.2007.17.
13. Závada J, Nytrová P, Wandinger KP et al. Seroprevalence and specificity of NMO-IgG (anti-aquaporin 4 antibodies) in patients with neuropsychiatric systemic lupus erythematosus. Rheumatol Int 2013; 33 (1): 259–263. doi: 10.1007/s00296-011-2176-4.
14. Salvarani C, Brown RD Jr, Calamia KT et al. Primary CNS vasculitis with spinal cord involvement. Neurology 2008; 70 (24 Pt 2): 2394–2400. doi: 10.1212/01.wnl.0000314687.69681.24.
15. Yadav N, Pendharkar H, Kulkarni GB. Spinal cord infarction: clinical and radiological features. J Stroke Cerebrovasc Dis 2018; 27 (10): 2810–2821. doi: 10.1016/j.jstrokecerebrovasdis.2018.06.008.
16. Štětkářová et al. Spinální neurologie. In: Štěkářová I, Nytrová P. Autoimunitní myelitidy. Praha: Maxdorf 2019: 284–310
17. Kumral E, Polat F, Güllüoglu H et al. Spinal ischaemic stroke: clinical and radiological findings and short-term outcome. Eur J Neurol 2011; 18 (2): 232–239. doi: 10.1111/j.1468-1331.2010.02994.x.
18. Freedman BA, Malone DG, Rasmussen PA et al. Surfer‘s myelopathy: a rare form of spinal cord infarction in novice surfers: a systematic review. Neurosurgery 2016; 78 (5): 602–611. doi: 10.1227/NEU.0000000000001089.
19. Dillen WL, Hendricks BK, Mannas JP et al. Surfer‘s myelopathy: a rare presentation in a teenage gymnast and review of the literature. J Clin Neurosci 2018; 50: 157–160. doi: 10.1016/j.jocn.2018.01.039.
20. Schroeder GD, Vaccaro AR. Cervical spine injuries in the athlete. Instr Course Lect 2017; 66: 391–402.
21. Keřkovský M, Šprláková-Puková A, Bednařík J et al.Význam MR zobrazení difuze míchy v diferenciální dia¬gnostice míšních lézí. Cesk Slov Neurol N 2013; 76/109 (4): 477–481.
22. Miyakis S, Lockshin MD, Atsumi T et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006; 4 (2): 295–306. doi: 10.1111/j.1538-7836.2006.01753.x.
23. Flanagan EP. Autoimmune myelopathies. HandbClin Neurol 2016; 133: 327–351. doi: 10.1016/B978-0-444-63432-0.00019-0.
24. Cavallieri F, Fini N, Contardi S et al. Subacute copper-deficiency myelopathy in a patient with occult celiac disease. J Spinal Cord Med 2017; 40 (4): 489–491. doi: 10.1080/10790268.2016.1246639.
25. Schwendimann RN. Metabolic and toxic myelopathies. Neurol Clin 2013; 31 (1): 207–218. doi: 10.1016/j.ncl.2012.09.002.
26. Weerasinghe D, Lueck C. Mimics and chameleons of optic neuritis. Pract Neurol 2016; 16 (2): 96–110. doi: 10.1136/practneurol-2015-001254.
27. Hickman SJ, Dalton CM, Miller DH et al. Management of acute optic neuritis. Lancet 2002; 360; 1953–1962. doi: 10.1016/s0140-6736 (02) 11919-2.
28. Chen JJ, Pittock SJ, Flanagan EP et al. Optic neuritis in the era of biomarkers. Surv Ophthalmol 2020; 65 (1): 12–17. doi: 10.1016/j.survophthal.2019.08.001.
29. Vanikieti K, Poonyathalang A, Jindahra P et al. Clinical characteristics and long-term visual outcome of optic neuritis in neuromyelitis optica spectrum disorder: a comparison between Thai and American-Caucasian cohorts. Mult Scler Relat Disord 2017; 17: 87–91. doi: 10.1016/j.msard.2017.07.013.
30. Zhao X, Qiu W, Zhang Y et al. A prospective case-control study comparing optical coherence tomography characteristics in neuromyelitis optica spectrum disorder – optic neuritis and idiopathic optic neuritis. BMC Ophthalmol 2018; 18 (1): 247. doi: 10.1186/s12886-018-0902-3.
31. Harding AE, Sweeney MG, Mil ler DH et al. Occurrence of a multiple sclerosis-like il lness in women whohave a Leber‘s hereditary optic neuropathy mitochondrial DNA mutation. Brain 1992; 115 (Pt 4): 979–989. doi: 10.1093/brain/115.4.979.
32. Kellar-Wood H, Robertson N, Govan GG et al. Leber’s hereditary optic neuropathy mitochondrial DNA mutations in multiple sclerosis. Ann Neurol 1994; 36 (1): 109–112. doi: 10.1002/ana.410360121.
33. Jansen PH, van der Knaap MS, de Coo IF. Leber‘s hereditary optic neuropathy with the 11,778 mtDNA mutationand white matter dis ease resembl ing multiple sclerosis: clinical, MRI and MRS fi ndings. J Neurol Sci 1996; 135 (2): 176–180. doi: 10.1016/0022-510x (95) 00287-c.
34. Petzold A, Plant GT. Chronic relapsing inflammatory optic neuropathy: a systematic review of 122 cases reported. J Neurol 2014; 261 (1): 17–26. doi: 10.1007/s00415-013-6957-4.
35. Wandinger KP, Stangel M, Witte T et al. Autoantibodies against aquaporin-4 in patients with neuropsychiatric systemic lupus erythematosus and primary Sjögren‘s syndrome. Arthritis Rheum 2010; 62 (4): 1198–1200. doi: 10.1002/art.27337.
36. Wang KC, Lee CL, Chen SY et al. Prominent brainstem symptoms/signs in patients with neuromyelitis optica in a Taiwanese population. J Clin Neurosci 2011; 18 (9): 1197–1200. doi: 10.1016/j.jocn.2010.12.052.
37. Shosha E, Dubey D, Palace J et al. Area postrema syndrome: frequency, criteria, and severity in AQP4-IgG-positive NMOSD. Neurology 2018; 91 (17): e1642–e1651. doi: 10.1212/WNL.0000000000006392.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2020 Číslo supplementum 1
Nejčtenější v tomto čísle
- Nálezy na magnetické rezonanci u neuromyelitis optica a onemocnění jejího širšího spektra
- Laboratorní vyšetření u neuromyelitis optica a onemocnění jejího širšího spektra
- Epidemiologie, klinický obraz a průběh onemocnění u neuromyelitis optica a onemocnění jejího širšího spektra
- Diferenciální diagnostika neuromyelitis optica a onemocnění jejího širšího spektra