
Novorozenecké záchvaty – současný pohled na problematiku
Neonatal seizures – current view of the issue
The newborn period poses the most vulnerable time in the development of epileptic seizures. The main predisposing factor is an increased neuronal excitability resulting from the incomplete maturation of a premature brain. From this point of view, premature newborns are at the highest risk of developing neonatal seizures. Early initiation of rational diagnostic and therapeutic intervention is often complicated by the vague or absent clinical manifestation of neonatal seizures. The variability of their clinical picture is reflected in the new draft of Neonatal Seizure Classification by International League Against Epilepsy (ILAE) from 2018. Timely diagnosis and initiation of adequate therapy with regard to etiology is, from the prognosis viewpoint, crucial. The strongest predictor of prognosis is etiology, as well as gestational age, initial findings during the neurological examination and ictal and interictal electroencephalographic features.
Keywords:
classification – therapy – neonatal seizures – EEG
Autoři:
K. Španělová; K. Česká; H. Ošlejšková; Š. Aulická
Působiště autorů:
Centrum pro epilepsie, Klinika dětské neurologie LF MU a FN Brno
Vyšlo v časopise:
Cesk Slov Neurol N 2020; 83(1): 48-56
Kategorie:
Přehledný referát
doi:
https://doi.org/10.14735/amcsnn202048
Souhrn
Novorozenecké období představuje nejvulnerabilnější periodu pro rozvoj epileptických paroxysmů. Zásadním predisponujícím faktorem je zvýšená neuronální excitabilita plynoucí z nedokončené maturace nezralého mozku. Z tohoto pohledu jsou nejrizikovější skupinou pro vznik novorozeneckých záchvatů nedonošení novorozenci. Včasné zahájení racionálních diagnosticko-terapeutických intervencí mnohdy komplikuje vágní až absentní klinická manifestace novorozeneckých záchvatů. Variabilitu jejich klinického obrazu reflektuje nová klasifikace novorozeneckých záchvatů dle Mezinárodní ligy proti epilepsii (International League Against Epilepsy; ILAE) z roku 2018. Včasné stanovení diagnózy a zahájení adekvátní terapie s ohledem na etiologii je z prognostického hlediska stěžejní. Nejsilnějším prognostickým prediktorem je etiologie, dále pak gestační stáří, vstupní nález při neurologickém vyšetření a EEG iktální a interiktální charakteristiky.
Úvod
Novorozenecké záchvaty (NZ) patří mezi nejčastější akutní neurologické stavy novorozeneckého období a jsou stále asociovány s vysokou mortalitou a morbiditou. Patofyziologickým podkladem hyperexcitačního stavu nezralého mozku je nedokončená maturace CNS. V tomto ohledu jsou nejvíce ohroženou skupinou nezralí novorozenci. Ačkoli většina záchvatů v novorozeneckém období jsou akutní symptomatické záchvaty (ASZ), malé procento NZ je projevem epilepsie. Dominující etiologií ASZ u donošených novorozenců je hypoxicko-ischemická encefalopatie (HIE). U nezralých novorozenců jsou za většinu epileptických paroxysmů zodpovědné cerebrovaskulární inzulty, tedy zejména intrakraniální hemoragie a mozkové infarkty. Idiopatické NZ (např. benigní familiární novorozenecké křeče) a novorozenecké epileptické syndromy (Ohtaharův syndrom, časná myoklonická epilepsie) tvoří jenom 2 % z celkového počtu. Spektrum etiologických faktorů vč. jejich poměrného zastoupení a obvyklého počátku manifestace NZ prezentuje obr. 1.
BFNS – benigní familiární neonatální záchvaty; BINC – benigní idiopatické neonatální křeče; HIE – hypoxicko-ischemická
encefalopatie
Fig. 1. Etiological factors of neonatal seizure development.
BFNS – benign familial neonatal seizure; BINC – benign idiopathic neonatal convulsions; HIE – hypoxic-ischemic encefalopathy
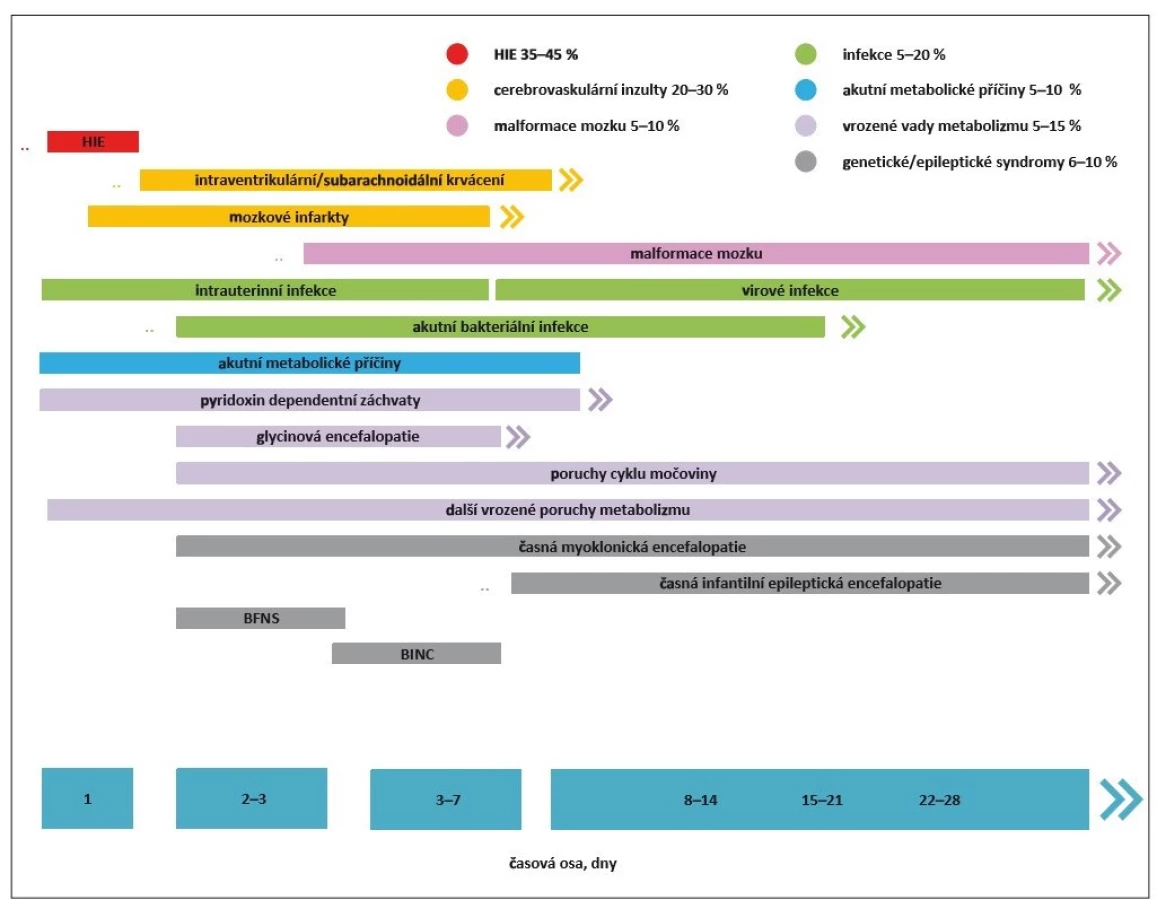
Incidence NZ se u donošených novorozenců pohybuje v rozmezí 1–4/ 1 000 živě narozených. U předčasně narozených je incidence vyšší a stoupá v závislosti na snižujícím se gestačním stáří a porodní hmotnosti. V kategorii novorozenců nízké porodní hmotnosti, tedy pod 2 500 g, je incidence 13,5/ 1 000 živě narozených. V kategorii velmi nízké porodní hmotnosti, tedy pod 1 500 g, je rozmezí velmi široké a pohybuje se mezi 55–130/ 1 000 živě narozených dětí. Incidence epilepsie se v prvním roce života u dětí, které prodělaly NZ, pohybuje v rozmezí 1–1,5/ 1 000 [1–5].
Patofyziologické aspekty
Mozek novorozence má unikátní fyziologické vlastnosti, které se v mnoha ohledech výrazně liší od neurofyziologie dospělých. Neuronální excitabilitu modulují věkově specifické mechanizmy na molekulární i buněčné úrovni, které v novorozeneckém období vedou k narušení stability klidového membránového potenciálu. Z toho vyplývá tendence nezralých neuronů v důsledku abnormální synchronní neuronální aktivity generovat epileptické paroxysmy. Tato vývojová regulace funkce buněčné membrány je patofyziologickým podkladem hyperexcitability [6–8].
Faktory determinující hyperexcitabilitu nezralého mozku:
- nezralost neurotransmisních systémů
- relativní nadbytek excitačních neurotransmiterů,
- nezralost inhibičních mechanizmů;
- věkově specifický energetický metabolizmus buňky;
- věkově vázaná exprese iontových kanálů;
- věkově vázaná modulace neuropeptidů;
- věkově vázaná časná aktivace mikroglie [5].
Díky intenzivní synaptogenezi a dendrifikaci probíhá velmi dynamický rozvoj neuronálních sítí a hustota synaptických spojů dosahuje v novorozeneckém období svého vrcholu. V časných stadiích postnatálního vývoje je zcela fyziologickým jevem přechodné zvýšení exprese excitačních receptorů pro glutamát, zejména NMDA (N-methyl-D-aspartát) a AMPA (α-amino-3-hydroxy-5-metyl 4-izoxazoleproprionát), méně pak kainátových receptorů. Vývojové regulaci podléhá také jejich podjednotkové složení, což ovlivňuje nejen funkční, ale také farmakologické vlastnosti receptoru. V nezralém mozku jsou hojně zastoupeny NMDA receptory obsahující převážně NR2B podjednotku. Tato vývojová modifikace podjednotkového složení receptorů prodlužuje trvání synaptické odpovědi v porovnání s NMDA receptory zralých neuronů, které jsou asociovány s NR2A podjednotkami [8,9]. Funkční vývojová modifikace AMPA receptorů spočívá ve snížené expresi GLUA2 podjednotky a vede ke zvýšení permeability pro vápenaté (Ca2+) ionty. Excitabilita je potencována také možnou paradoxně excitační odpovědí GABA (γ-amino-butyrát) receptorů způsobenou zpožděnou expresí KCC2 chloridového kotransportéru, který je spolu s NKCC1 kotransportérem zodpovědný za udržování homeostázy chloridových iontů. Převaha kotransportéru NKCC1 vede k vysoké koncentraci intracelulárního chloridu a následné depolarizaci. Svoji roli hraje také snížená exprese alfa1 podjednotky GABA receptorů. Vývojové regulaci podléhá také HCN iontový kanál, jehož izoforma HCN1 je v nezralém mozku sníženě exprimována, a tím je potencována dendritická excitabilita. Kompozice GABA receptorů nezralých neuronů částečně vysvětluje rezistenci na konvenční antiepileptika (anti-epileptic drugs; AED), která fungují jako agonisté GABA [8,10].
Kompozice receptorů nezralých neuronů v porovnání se zralými neurony:
- NR2B > NR2A podjednotka NMDA receptorů;
- GLUA1 > GLUA2 podjednotka AMPA receptorů;
- NKCC1 > KCC2 kotransportér GABA receptorů;
- non-alfa1 > alfa1 podjednotka GABA receptorů.
Neuromodulačním peptidem, který potencuje neuronální excitabilitu, je kortikoliberin (corticotropin-releasing hormone; CRH), který je spolu se svými receptory v neonatálním období zvýšeně exprimován [8,11]. V patogenezi epileptických paroxysmů nelze opomenout úlohu mikroglie, která se v průběhu epileptogeneze podílí na modulaci stability membránového potenciálu. Jedná se o velmi heterogenní buněčnou populaci, která vykazuje velkou regionální a věkově determinovanou funkční variabilitu [12–14]. V závislosti na konkrétních podnětech mikroprostředí, lokalizaci a věku jsou mikroglie fenotypicky odlišné a mohou mít role neuroprotektivní nebo naopak neurotoxické [15].
Ovlivnění neuronální funkce
Recentní studie podporují fakt, že epileptické paroxysmy samy o sobě mají negativní dopad na vývoj mozku a mohou vést k neurologickému poškození dítěte. Míra poškození a s ním asociovaných následků se odvíjí primárně od etiologie NZ, která je nejdůležitějším prognostickým indikátorem. Do určité míry koreluje s délkou trvání záchvatu, recidivami a vývojovým stupněm CNS. Je tedy zřejmé, že záchvaty, zejména refrakterní, prolongované až status epilepticus (SE), které se manifestují velmi časně postnatálně, ovlivňují buněčnou morfologii i fyziologii a narušují neuronální funkce. Klinické konsekvence vyplývají z vlivu NZ především na epileptogenezi, kognitivní a behaviorální funkce [16–18]. Vyvíjející se mozek je ve srovnání s mozkem dospělého jedince odolnější vůči poškození indukovanému záchvatem. Odolnějším jej činí nezralost biochemických kaskád zodpovědných za indukci apoptózy, vysoké koncentrace neuroprotektivně působícího faktoru BDNF82, nižší hladiny cytokinů v rámci zánětlivé odpovědi indukované záchvatem a nižší míra oxidativního stresu. V prvních postnatálních týdnech je také stabilnější zachování syntézy GABA v průběhu prolongovaných záchvatů a SE. Stabilita syntézy GABA v hipokampu spolu s nezralou myelinizací a nižší hustotou aktivních synapsí znesnadňuje šíření epileptiformní aktivity a lze tak vysvětlit převážně fokální nebo multifokální povahu záchvatů u novorozenců [19].
Klinická manifestace a EEG rysy
Většina paroxysmálních projevů novorozeneckého období je neepileptické geneze. Řada z nich jsou zcela benigní příhody (např. benigní spánkové myoklonie), zatímco některé, i přes svou neepileptickou patogenezi, souvisí s neurologickým onemocněním dítěte (např. hyperekplexie, apnoické pauzy, dystonické ataky). Známky asociované převážně s neepileptickou patogenezí paroxysmálního projevu jsou generalizované klinické projevy, absence EEG korelátu (absence však epileptický původ zcela nevylučuje – elektroklinická disociace) a přerušení záchvatu manipulačními manévry [6,20].
Více než dvě třetiny NZ se manifestují obvykle již v prvním týdnu života. Díky nezralosti CNS se epileptické paroxysmy v neonatálním období a v populaci starších dětí a dospělých liší řadou klinických a elektrografických rysů. Variabilní a v řadě případů diskrétní až absentní klinický obraz NZ znesnadňuje diagnostiku a oddaluje včasné zahájení racionální terapie. Situace, kdy dochází ke kombinaci epileptických a neepileptických paroxysmů, nejsou výjimečné a semiologie některých iktálních a postiktálních symptomů může být velmi snadno zaměněna za normální behaviorální, motorické a autonomní projevy novorozence [4,5,19].
Stávající definice záchvatů je založena na detekci příznaků nebo abnormálního chování pacienta v průběhu epileptického paroxysmu. Záchvat je definován jako přechodný výskyt symptomů vznikajících v důsledku abnormální synchronní (epileptické) neuronální aktivity v mozku [7,20,21]. Tato definice ovšem nezohledňuje celé spektrum NZ. Na základě elektro-klinické korelace rozlišujeme v neonatálním období následující kategorie:
- klinický záchvat – klinický projev nekoreluje se simultánním EEG nálezem;
- elektroklinický záchvat – klinický projev koreluje se simultánním EEG nálezem;
- elektrografický záchvat – EEG nález nemá klinický korelát (subklinický, nonkonvulzivní, okultní).
V EEG obraze se NZ vyznačují délkou trvání ≥10 s, náhlým a zřetelným počátkem a koncem, mají fokální původ s šířením a typický je vývoj amplitudy a opakovací frekvence. Je třeba zdůraznit, že jakákoliv rytmická aktivita v EEG u novorozenců je velmi suspektní. Jestliže máme k dispozici minimálně 30min EEG záznam, ve kterém je > 50 % záznamu tvořeno záchvatovou aktivitou, pak se jedná o SE. Elektrografický záchvat lze definovat jako kontinuální nebo periodicky nastupující rytmický paroxysmální vzorec s minimální voltáží 2 μV v délce trvání nejméně 10 s. Paroxysmální interiktální rytmické změny EEG s nebo bez vývoje trvající méně než 10 s, tzv. BIRDs (brief interictal rythmic discharges), jsou asociovány se zvýšeným rizikem abnormálního neurologického vývoje a mohou predikovat záchvaty v aktuálním nebo následném EEG. Dle některých autorů lze BIRDs s vývojem považovat za velmi krátké elektrografické záchvaty, zatímco BIRDs bez vývoje mohou sloužit jako prognostický indikátor a prediktor záchvatů [21–23]. Elektrografické záchvaty se vyskytují zejména u encefalopatických a kriticky nemocných novorozenců. V populaci novorozenců postižených HIE tvoří elektrografické záchvaty 50–80 %. Mohou se objevit po podání myorelaxancií a některých antiepileptik, zejména fenobarbitalu, kdy dochází pouze k potlačení klinické manifestace, ale přetrvává elektrografická záchvatová aktivita – elektroklinická disociace [21,23,24].
Epileptické paroxysmy v populaci nedonošených novorozenců v porovnání s donošenými vykazují některé společné elektrografické vlastnosti, které odráží úroveň maturace CNS. Jedná se o kratší dobu trvání, pomalejší frekvence, menší zónu počátku záchvatů a vzácněji dochází k propagaci iktální EEG aktivity. Epileptogenní zóna je častěji lokalizována v oblasti zadních kvadrantů [25].
Nová klasifikace
Doposud užívané klasifikace NZ byly složité, nejednotné a zahrnovaly i záchvatové projevy neepileptické geneze. Jejich interpretace tedy naráží na řadu úskalí. Např. klasifikační schéma NZ dle Volpe vychází pouze z klinické manifestace záchvatů (klonické, tonické, myoklonické a subtilní záchvaty) [6]. Klasifikace NZ dle Mizrahi a Kellaway (fokální tonické, fokální klonické, myoklonické, spazmy, elektrografické, generalizované tonické záchvaty a motorické automatizmy) [26] zohledňuje také patofyziologii záchvatu.
Nová klasifikace záchvatů částečně reflektuje recentně revidovanou novou klasifikaci záchvatů International League Against Epilepsy (ILAE; 2017) a rozdělení NZ podstatně zjednodušuje (tab. 1). Cílem je přinést systém, který bude snadno implikovatelný do klinické praxe a srozumitelný širokému oborovému spektru. Změny jsou založeny na EEG verifikaci záchvatů, zjednodušení terminologie skutečně epileptických záchvatů a zavedení nových pojmů, které popisují záchvaty typické, resp. nejčastěji se vyskytující právě v novorozeneckém věku. Čistě klinickou evaluací jsme schopni zachytit přibližně polovinu záchvatů, pouze však za předpokladu prakticky kontinuální monitorace pacienta. S ohledem na stoupající incidenci elektrografických záchvatů u novorozenců je EEG monitorace stěžejní diagnostickou metodou. Zlatým standardem je video EEG monitorace, nicméně nelze opomenout význam amplitudové elektroencefalografie (aEEG). Benefity aEEG lze spatřovat v možnosti kontinuální bed-side monitorace, relativně snadné přístrojové aplikaci a interpretaci záznamu. Metoda umožňuje detekci SE, suppression burst vzorce a jsme schopni sledovat dlouhodobé změny mozkové aktivity, což přispívá k predikci neurologického vývoje. Naproti tomu relativní jednoduchost aEEG vede k jisté redukci informace a není možné zachytit záchvaty trvající < 30 s, jakými jsou např. epileptické spazmy nebo myoklonické záchvaty. Problematická je detekce fokálních abnormit a záchvatů probíhajících distálně od elektrod [27,28].
![Klasifikace novorozeneckých záchvatů dle klinických projevů [4].](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image_pdf/92b38640fddc49a25f4d4bde7f564e6e.png)
Po verifikaci epileptické patogeneze je nutné určit, zda se jedná o záchvat s klinickými projevy nebo bez nich. Přesná lokalizace zóny počátku záchvatu nemá u většiny dětí s NZ praktický význam pro volbu léčebného postupu, ojediněle se však i v této věkové kategorii můžeme setkat s kandidáty časné epileptochirurgie. Stejně tak nelze bez EEG monitorace validně zhodnotit stav vědomí u novorozenců [20,22]. Nová klasifikace NZ dle ILAE 2018 zahrnuje typy záchvatů relevantní pro tuto věkovou kategorii. Konkrétní typ záchvatu je určen podle dominujícího klinického příznaku, nikoliv podle prvního klinického příznaku. V některých případech je obtížné určit dominující klinický příznak, proto byl zaveden nový typ záchvatu – sekvenční, pro který je charakteristický sled příznaků nebo projevů často s měnlivou lateralizací. Elektrograficky lze detekovat překrývání iktálních vzorců, jejich vývoj a změny v čase. Sekvenční záchvaty se vyskytují často v asociaci s genetickou nebo metabolickou etiologií a také u benigní familiární neonatální epilepsie (BFNE). Naopak záchvaty emoční, senzorické, kognitivní a atonické, které popisuje klasifikace dle ILAE 2017, nelze v neonatálním období diagnostikovat, proto je nová klasifikace neuvádí. Samostatnou skupinu reprezentuje kategorie záchvatů elektrografických [29]. Diagnostický algoritmus vč. klasifikace NZ je graficky zpracován na obr. 2.
aEEG – amplitudová EEG; V-EEG – video-EEG
Fig. 2. Diagnostic algorithm and neonatal seizure classification by the International League
Against Epilepsy 2018 [43].
aEEG – amplitude-integrated EEG; V-EEG – video-EEG
![Diagnostický algoritmus a klasifikace novorozeneckých záchvatů dle International
League Against Epilepsy 2018 [43].<br>
aEEG – amplitudová EEG; V-EEG – video-EEG<br>
Fig. 2. Diagnostic algorithm and neonatal seizure classification by the International League
Against Epilepsy 2018 [43].<br>
aEEG – amplitude-integrated EEG; V-EEG – video-EEG](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image_pdf/ec57ffba0f1dab4450c9abdf47df0db3.jpeg)
Správné určení typu záchvatu podporuje stanovení etiologické diagnózy a ovlivňuje tak diagnosticko-terapeutický algoritmus. Skupina autorů klasifikace NZ ILAE 2018 si všimla určité asociace mezi iktální semiologií a etiologií epileptických paroxysmů. Konkrétně genetická etiologie je ve většině případů spojena se záchvaty tonickými a/ nebo sekvenčními, elektrografické záchvaty vídáme u nezralých novorozenců bez ohledu na etiologii a dále ve spojení s HIE a infekčními příčinami záchvatů. Následkem CMP vznikají nejčastěji fokální klonické záchvaty a dědičné poruchy metabolizmu jsou mnohdy spojeny se záchvaty myoklonickými. Časné odlišení ASZ od neonatálních epilepsií má významné terapeutické a prognostické konsekvence [29] (obr. 3).
Fig. 3. Epilepsy classification by the International League Against Epilepsy 2018 [43].
![Klasifikace epilepsií dle International League Against Epilepsy 2018 [43].<br>
Fig. 3. Epilepsy classification by the International League Against Epilepsy 2018 [43].](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image_pdf/a23fa447c24aa686dd3d09f9ff77c8f5.png)
Díky významnému technologickému pokroku v oblasti genetické analýzy roste podíl geneticky determinovaných epilepsií. Stanovení genetické diagnózy umožňuje identifikovat potenciálně léčitelné epilepsie, např. syndrom GLUT1 deficience (SCL2A1), kde je metodou volby ketogenní dieta, a také může poskytnout velmi přesné prognostické informace. Dosud bylo definováno více než 100 genů asociovaných s epileptickými encefalopatiemi s časným počátkem. Protože klinické projevy infantilních epileptických syndromů se často překrývají a mají heterogenní genetické příčiny, představují metody masivně paralelního sekvenování (next generation sequencing; NGS) silnější diagnostický nástroj než testování jednoho genu. V klinické praxi patří k běžně užívaným metodám NGS vyšetření panelů epileptických genů celoexomové sekvenování nebo celogenomové sekvenování. Sangerovo sekvenování je přednostně indikováno u pacientů s dobře definovanými fenotypy asociovanými s jedinou genovou mutací a je zlatým standardem pro detekci malých sekvenčních variant. Genetické příčiny neonatálních epilepsií spadají do kategorií, jejichž přehled je spolu s konkrétními příklady uveden v tabulce v tab. 2 [5,30].
![Genetické příčiny neonatálních epilepsií [30].](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image_pdf/c4bc3268a3424ef8a27d563999d8c5f9.png)
Epileptické syndromy novorozeneckého období
Fenotypové spektrum novorozeneckých epilepsií se pohybuje od benigních neonatálních záchvatů až po devastující epileptické encefalopatie [30].
Benigní/„self-limiting“ („se spontánníremisí“) neonatální záchvaty se řadí mezi prognosticky příznivé epileptické syndromy nejasné etiologie. Manifestace je velmi časná, typicky mezi 4.–6. postnatálním dnem se u fyziologického novorozence objeví multifokální klonické záchvaty s apnoickou pauzou nebo bez apnoické pauzy. Nekonstantně je možný přechod až do unilaterálního SE [7].
Benigní/ „self-limiting“ („se spontánní remisí“) familiární neonatální epilepsiepředstavuje vzácné autozomálně dominantně dědičné onemocnění způsobené mutací genů kódujících draslíkové kanály – KCNQ2 (většina případů v lokalizaci 20q13.3, méně případů KCNQ3 v lokalizaci 8q24) [30]. Vyskytují se obvykle 2.–3. den a klinicky se manifestují jako multifokální klonické, vzácně tonické záchvaty, nekonstantně s apnoickými pauzami nebo event. s vokalizací. Prognóza je velmi příznivá a v průběhu několika měsíců lze očekávat spontánní remisi, proto je otázka terapie kontroverzní [31]. Asi 15 % pacientů s BFNNC je ohroženo pozdějším rozvojem epilepsie. Samostatným geneticky odlišným syndromem jsou benigní familiární neonatální a infantilní záchvaty, které se typicky manifestují na přelomu novorozeneckého a kojeneckého období s remisí do 1 roku a jsou asociovány s mutací v genu SCN2A [32].
Iktální EEG nálezy u těchto dvou neonatálních epileptických syndromů vykazují interindividuální variabilitu. Interiktální EEG záznam může být normální, abnormální fokální nebo s projevy multifokality. U více než poloviny pacientů detekujeme v interiktálním EEG záznamu „theta point alternant“ [7].
Časná myoklonická encefalopatie (early myoclonic encephalopathy; EME) a časná infantilní epileptická encefalopatie (early-infantile epileptic encephalopathy; EIEE) – Ohtahara syndrom, jsou prognosticky nepříznivé epileptické encefalopatie, které dle mnohých autorů reprezentují kontinuum a jejichž patofyziologický podklad se překrývá. Nejčastější patogenní mutace zahrnují STXBP1, PNKP, KCNQ2, SCN2A, méně často identifikované kauzální geny jsou PNPO, PIGA, ARX (muži), CASK (muži) a SIK1. Existují popsané jednotlivé kazuistiky časných epileptických encefalopatií různého fenotypového spektra, kde byly popsány patogenní varianty CDKL5, SLC25A22, ALG1, GABRA1, SEPSECS a SLC1A2 [30,33].
Časná myoklonická encefalopatie bývá obvykle asociována s dědičnými metabolickými poruchami (non-ketotická hyperglycinemie, vitamin-dependentní epilepsie, mitochondriální a peroxisomální poruchy apod.) nebo s malformacemi kortikálního vývoje. Klinicky se manifestuje v průběhu prvního měsíce života (někdy již několik hodin po narození) fokálními eratickými myokloniemi, méně často vídáme myoklonie generalizované. Tonické záchvaty jsou vzácné. EEG obraz vykazuje rysy suppression burst vzorce s akcentací ve spánku a u poloviny pacientů se EEG záznam vyvíjí v průběhu několika měsíců směrem k diskontinuální hypsarytmii. Všichni pacienti jsou postiženi těžkou psychomotorickou retardací a téměř polovina umírá před dosažením 2. roku věku [7,33]. EIEE je většinou podmíněna malformacemi CNS. K manifestaci dochází obvykle během prvních 2 týdnů života. Pro EIEE jsou charakteristické záchvaty tonické, konkrétně symetrické i asymetrické tonické flekční spazmy izolované nebo v sériích nezávisle na spánku. Zpravidla se neobjevují myoklonie, mohou se vyskytnout fokální motorické záchvaty, hemikonvulze nebo generalizované tonicko-klonické záchvaty. Pro EEG záznam je typický suppression burst vzorec, přičemž v průběhu záchvatu může dojít k desynchronizaci. S věkem obvykle dochází k přechodu do Westova a následně do Lennox-Gastautova syndromu [7,31,33].
Terapie
Terapeutická strategie se za ideálních okolností odvíjí od etiologie NZ. Jejím cílem je zabránit rozvoji ireverzibilních neurologických změn, minimalizovat riziko rekurence záchvatů a zajistit tak pacientovi co nejlepší prognózu. Kauzální terapie, je-li to vzhledem k základnímu onemocnění možné, je neodmyslitelnou součástí terapeutického postupu a probíhá paralelně se symptomatickou terapií [34].
Zavádění nových léčebných strategií v posledních letech vedlo ke zlepšení prognózy pacientů s HIE. Časně indikovaná a správně vedená terapeutická hypotermie má prokazatelně neuroprotektivní efekt a snižuje riziko následného rozvoje epilepsie [35].
Metabolické příčiny vyžadují korekci poruch vnitřního prostředí ve smyslu iontové rovnováhy, acidobazické rovnováhy, udržování normoglykemie a normovolemie. Pyridoxin dependentní epilepsie vyžadují intravenózní podání pyridoxinu v dávce 100 mg a následné podávání 15–30 mg/ kg/ den ve třech dílčích dávkách. Pyridoxal-fosfát dependentní epilepsie jsou na terapii pyridoxinem refrakterní, ale reagují na léčbu pyridoxal-fosfátem (30 mg/ kg/ den pyridoxal-fosfátu a 3–5 mg/ kg/ den leucovorinu) [36–38].
Antiepileptická terapie
Nezralost neurotransmisních systémů novorozenců často komplikuje léčbu konvenčními antiepileptiky. Ve srovnání s populací starších dětí se terapie liší. Výběr konkrétního antiepileptika se odvíjí od výchozího stavu pacienta, jeho kardiopulmonální stability a hepatálních a renálních funkcí. Racionálním krokem je zvolit k akutní léčbě takové antiepileptikum případně kombinace antiepileptik, které lze následně podávat i v rámci udržovací terapie.
Lékem 1. volby u NZ je fenobarbital. Mimo antikonvulzivního efektu má prokázané neuroprotektivní vlastnosti. Podává se v úvodním bolusu 20–30 mg/ kg i.v. rychlostí < 5 mg/ kg/ min. Jestliže je terapie efektivní, pokračujeme dávkou 4–6 mg/ kg/ den ve dvou dílčích dávkách. Fenobarbital je metabolizován hepatorenální cestou, proto je nutná obezřetnost v případě poruchy funkce jater nebo ledvin, kdy mohou být standardní dávky pro pacienta toxické. V případě neúspěchu fenobarbitalu volíme z řady antiepileptik 2. volby, mezi které patří fenytoin, midazolam, klonazepam, levetiracetam event. lidokain. Pro úplnost je třeba zmínit také topiramát. Fenytoin představuje svým mechanizmem účinku alternativu fenobarbitalu. Jeho hlavní nevýhodou je kolísání hladiny v krvi. Jeho nežádoucími účinky mohou být arytmie a hypotenzní efekt. Podává se v úvodní nasycovací dávce 20 mg/ kg i.v. rychlostí max. 3 mg/ kg/ min, následně 4–6 mg/ kg/ den ve dvou dílčích dávkách. Midazolam je krátce působící benzodiazepin, který se používá zejména u refrakterního SE. Podává se v úvodním bolusu 0,15 mg/ kg a pokračujeme v kontinuální infuzi v dávce 1 μg/ kg/ min s možnou titrací dle efektu a tolerance. Alternativu midazolamu představuje klonazepam, který se používá v úvodním bolusu 0,01–0,02 mg/ kg i.v., následně pokračujeme kontinuální infuzí 0,1 mg/ kg/ den. Lidokain lze uplatnit u refrakterních NZ. K jeho nežádoucím účinkům patří poruchy srdečního rytmu. Úvodní bolusová dávka je 2 mg/ kg/ 10 min následovaná kontinuální infuzí 7 mg/ kg/ h (po 4 h snižovat dávku o 50 %). Poslední desetiletí je spjato se zavedením nových antiepileptik do běžné praxe, přičemž některé z nich byly v „off label“ indikaci použity k léčbě NZ – levetiracetam a topiramát. Levetiracetam má velmi příznivý farmakokinetický profil a je velmi dobře tolerován. Nasycovací dávka je 40 mg/ kg i.v., následně lze pokračovat dávkou 40–60 mg/ kg/ den ve 2–3 dílčích dávkách. Topiramát má významné neuroprotektivní vlastnosti zejména u NZ asociovaných s HIE. V současné době ovšem není k dispozici ve formě pro parenterální podání. Epilepsie na podkladě tuberózní sklerózy je v novorozeneckém věku vzácná, nicméně zejména u pacientů s prenatálně diagnostikovaným nálezem rhabdomyomů srdce je nutné dlouhodobé sledování a časné podchycení rozvoje epilepsie. Terapeuticky v tomto případě upřednostňujeme vigabatrin [37].
Prognóza
Nejsilnějším prognostickým prediktorem je etiologie, jejíž charakter determinuje závažnost následného postižení pacienta. V porovnání s jinými příčinami bývá nepříznivá prognóza obvykle asociována s HIE, krvácením do mozku, infekcemi a malformacemi CNS [39]. Dalšími faktory, které ovlivňují prognózu pacienta, jsou gestační stáří, vstupní nález při neurologickém vyšetření a elektrografické iktální a interiktální charakteristiky [40]. Vyšší riziko horších následků mají novorozenci s trvale abnormním pozadím základní aktivity dle EEG, novorozenci se záchvaty rezistentními k antikonvulzivní terapii a novorozenci s nízkým Apgar skóre [34]. Mortalita zejména v rizikových populacích novorozenců a v asociaci s HIE se pohybuje kolem 15–20 % [2].
Recentní poznatky ukazují, že 8–43 % NZ se může vyvinout do SE. Díky studii francouzských autorů zabývajících se hodnocením prediktorů rozvoje SE u novorozenců se podařilo identifikovat dva nezávislé rizikové faktory. Jedná se o závažný nález při vstupním neurologickém vyšetření a hypoglykemii při narození. Normální postiktální klinické vyšetření bylo hodnoceno jako prognosticky příznivý faktor, ale pouze u novorozenců s izolovanými NZ. S délkou trvání SE se zvyšuje riziko závažných následků, proto bylo navrženo zkrácení arbitrární časové hranice pro neonatální SE z 30 na 15 min [41].
K závažným následkům NZ patří dětská mozková obrna, psychomotorická retardace, mentální deficit a následný rozvoj epilepsie. Epilepsií je postiženo 18–25 % dětí s NZ, přičemž u více než poloviny pacientů dochází k její manifestaci již v prvním roce života. Její následný rozvoj negativně potencuje řada faktorů – nízká porodní hmotnost, abnormální neuroradiologický nález, refrakterní záchvaty nebo SE, trvale abnormní EEG pozadí, multifokální EEG nález nebo kontralaterální propagace epileptické aktivity [40,42].
Grantová podpora
Tento projekt byl podpořen z fondu LF MU juniorský výzkumník Štefania Aulická. Dále podpořeno projektem MZČR-RVO (FN Brno, 65269705).
Konflikt zájmů
Autoři deklarují, že v souvislosti s předmětem studie nemají žádný konflikt zájmů.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Přijato k recenzi: 15. 10. 2019
Přijato do tisku: 13. 1. 2020
MUDr. Štefánia Aulická, Ph.D.
Centrum pro epilepsie
Klinika dětské neurologie
LF MU a FN Brno
Černopolní 9
613 00 Brno
e-mail: stefania.aulicka@gmail.com
Zdroje
1. Vasudevan C, Levene M. Epidemiology and aetiology of neonatal seizures. Semin Fetal Neonatal Med 2013; 18(4): 185–191. doi: 10.1016/ j.siny.2013.05.008
2. Glass HC, Shellhaas RA, Wusthoff CJ et al. Contemporary profile of seizures in neonates: a prospective cohort study. J Pediatr 2016; 174: 98–103. doi: 10.1016/ j.jpeds.2016.03.035
3. Uria-Avellanal C, Marlow N, Rennie JM. Outcome following neonatal seizures. Semin Fetal Neonatal Med 2013; 18(4): 224–232. doi: 10.1016/ j.siny.2013.01.002
4. Boylan GB, Pressler RM, Rennie JM et al. Outcome of electroclinical, electrographic, and clinical seizures in the newborn infant. Devl Med Child Neurol 2007; 41(12): 819–825. doi: 10.1017/ s0012162299001632.
5. Shellhaas RA, Wusthoff CJ, Tsuchida TN et al. Profile of neonatal epilepsies: characteristics of a prospective US cohort. Neurology 2017; 89(9): 893–899. doi: 10.1212/ WNL.0000000000004284
6. Abend NS, Jensen FE, Inder TE et al. Neonatal Seizures. In: Volpe’s neurology of the newborn. Amsterdam: Elsevier 2018: 275–321.
7. Pressler RM. Neonatal seizures. [online]. Available from URL: https:/ / www.epilepsysociety.org.uk/ sites/ default/ files/ attachments/ Chapter06Pressler2015.pdf.
8. Jensen FE. Neonatal seizures: an update on mechanisms and management. Clin Perinatol 2009; 36(4): 881–900. doi: 10.1016/ j.clp.2009.08.001
9. Jiang Q, Wang J, Wu X et al. Alterations of NR2B and PSD-95 expression after early-life epileptiform discharges in developing neurons. Int J Dev Neurosci 2007; 25(3): 165–70. doi: 10.1016/ j.ijdevneu.2007.02.001
10. Glass HC. Neonatal seizures: advances in mechanisms and management. Clin Perinatol 2014; 41(1): 177–190. doi: 10.1016/ j.clp.2013.10.004
11. Hooper A, Fuller PM, Maguire J. Hippocampal corticotropin-releasing hormone neurons support recognition memory and modulate hippocampal excitability. PLOS ONE 2018; 13(1): e0191363. doi: 10.1371/ journal.pone.0191363
12. Eyo UB, Murugan M, Wu LJ. Microglia-neuron communication in epilepsy. Glia 2017; 65(1), 5–18. doi: 10.1002/ glia.23006.
13. Koh S. Role of neuroinflammation in evolution of childhood epilepsy. J Child Neurol 2018; 33(1): 64–72. doi: 10.1177/ 0883073817739528
14. Ritzel RM, Patel AR, Pan S et al. Age- and location-related changes in microglial function. Neurobiol Aging 2015; 36(6): 2153–2163. doi: 10.1016/ j.neurobiolaging.2015.02.016.
15. Hiragi T, Ikegaya Y, Koyama R. Microglia after seizures and in epilepsy. Cells 2018; 7(4). pii: E26. doi: 10.3390/ cells7040026.
16. Ben-Ari Y, Holmes GL. Effects of seizures on developmental processes in the immature brain. Lancet Neurol 2006; 5(1): 1055–1063. doi: 10.1016/ S1474-4422(06)70626-3.
17. Nardou R, Ferrari DC, Ben-Ari Y. Mechanisms and effects of seizures in the immature brain. Semin Fetal Neonatal Med 2013; 18(4): 175–184. doi: 10.1016/ j.siny.2013.02.003.
18. Holmes GL. Effect of seizures on the developing brain and cognition. Semin Pediatr Neurol 2016; 23(2): 120–126. doi: 10.1016/ j.spen.2016.05.001.
19. Sankar R, Shin DH, Wasterlain CG. GABA metabolism during status epilepticus in the developing rat brain. Brain Res Dev Brain Res 1997; 98(1): 60–64. doi: 10.1016/ S0165-3806(96)00165-4.
20. Panayiotopoulos CP. The epilepsies: seizures, syndromes and management. 2nd edition. In: Panayiotopoulos CP. Chapter 5: Neonatal seizures and neonatal syndromes. Chipping Norton, England: Bladon Medical Publishing 2005.
21. Scheffer IE, Berkovic S, Capovilla G et al. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017; 58(4): 512–521. doi: 10.1111/ epi.13709.
22. Nagarajan L, Palumbo L, Ghosh S. Brief electroencephalography rhythmic discharges (BERDs) in the neonate with seizures: their significance and prognostic implications. J Child Neurol 2011; 26(12): 1529–1533. doi: 10.1177/ 0883073811409750.
23. Oliveira AJ, Nunes ML, Haertel LM et al. Duration of rhythmic EEG patterns in neonates: new evidence for clinical and prognostic significance of brief rhythmic discharges. Clin Neurophysiol 2000; 111(9): 1646–1653. doi: 10.1016/ s1388-2457(00)00380-1.
24. Boylan GB, Rennie JM, Pressler RM et al. Phenobarbitone, neonatal seizures, and video-EEG. Arch Dis Child Fetal Neonatal Ed 2002; 86(3): F165–F170. doi: 10.1136/ fn.86.3.F165
25. Janáčková S, Boyd S, Yozawitz E et al. Electroencephalographic characteristics of epileptic seizures in preterm neonates. Clin Neurophysiol 2016; 127(6): 2721–2727. doi: 10.1016/ j.clinph.2016.05.006
26. Mizrahi EM, Kellaway P. Characterization and classification of neonatal seizures. Neurology 1987; 37(12): 1837–1844. doi: 10.1212/ WNL.37.12.1837
27. Fernández IS, Loddenkemper T. aEEG and cEEG: Two complementary techniques to assess seizures and encephalopathy in neonates: editorial on “Amplitude-integrated EEG for detection of neonatal seizures: a systematic review” by Rakshasbhuvankar et al. Seizure 2015; 33: 88–89. doi: 10.1016/ j.seizure.2015.10.010
28. Pisani F, Pavlidis E. The role of electroencephalogram in neonatal seizure detection. Expert Rev Neurother 2018; 18(2): 95–100. doi: 10.1080/ 14737175.2018.1413352
29. International League Against Epilepsy. Neonatal seizure classification. [online]. Available from URL: https:/ / www.ilae.org/ files/ dmfile/ NeonatalSeizureClassification-ProofForWeb.pdf.
30. Axeen EJT, Olson HE. Neonatal epilepsy genetics. Sem Fetal Neonatal Med 2018; 23(3): 197–203. doi: 10.1016/ j.siny.2018.01.003
31. Komárek V. Léčba epileptických syndromů u dětí. Cesk Slov Neurol N 2007; 70/ 103(5): 473–485.
32. Poduri A, Lowenstein D. Epilepsy genetics – past, present, and future. Curr Opin Genet Dev 2011; 21(3): 325–332. doi: 10.1016/ j.gde.2011.01.005.
33. Beal JC, Cherian K, Moshe SL. Early-onset epileptic encephalopathies: Ohtahara syndrome and early myoclonic encephalopathy. Pediatr Neurol 2012; 47(5): 317–323. doi: 10.1016/ j.pediatrneurol.2012.06.002.
34. Tadic BV, Kravljanac R, Sretenovic V et al. Long--term outcome in children with neonatal seizures: a tertiary center experience in cohort of 168 patients. Epilepsy Behav 2018; 84: 107–113. doi: 10.1016/ j.yebeh.2018.05.002.
35. Inoue T, Shimizu M, Hamano S et al. Epilepsy and West syndrome in neonates with hypoxic-ischemic encephalopathy. Pediatr Int 2014; 56(3): 369–372. doi: 10.1111/ ped.12257.
36. Shellhaas R. Treatment of neonatal seizures. [online]. Available from URL: https:/ / www.uptodate.com/ contents/ treatment-of-neonatal-seizures.
37. Aulická Š, Aulický P, Česká K et al. Generalizovaný konvulzivní status epilepticus v dětském věku. Anest intenziv Med 2018; 29(3): 139–147.
38. Yozawitz E, Stacey A, Pressler RM. Pharmacotherapy for seizures in neonates with hypoxic ischemic encephalopathy. Paediatr Drugs 2017; 19(6): 553–567. doi: 10.1007/ s40272-017-0250-4.
39. Kang SK, Kadam SD. Neonatal seizures: impact on neurodevelopmental outcomes. Front Pediatr 2015; 3: 101. doi: 10.3389/ fped.2015.00101.
40. Pisani F, Spagnoli C. Neonatal seizures: a review of outcomes and outcome predictors. Neuropediatrics 2016; 47(1): 12–19. doi: 10.1055/ s-0035-1567873.
41. Gokce-Samar Z, Ostrowsky-Coste K, Gauthier-Morel D et al. Predictive factors and prognostic value for status epilepticus in newborns. Eur J PaediatrNeurol 2019; 23(2): 270–279. doi: 10.1016/ j.ejpn.2019.01.006.
42. Buraniqi E, Sansevere AJ, Kapur K et al. Electrographic seizures in preterm neonates in the neonatal intensive care unit. J Child Neurol 2017; 32(10): 880–885. doi: 10.1177/ 0883073817713918.
43. The ILAE Classification of Seizures & the Epilepsies: Modification for Seizures in the Neonate. Proposal from the ILAE Task Force on Neonatal Seizures. [online]. Available from URL: https:/ / www.ilae.org/ files/ dmfile/ NeonatalSeizureClassification-ProofForWeb.pdf
Štítky
Dětská neurologie Neurochirurgie Neurologie Pediatrie Praktické lékařství pro děti a dorostČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2020 Číslo 1
Nejčtenější v tomto čísle
- Novorozenecké záchvaty – současný pohled na problematiku
- Možnosti prevence Alzheimerovy choroby
- Primární non-Hodgkinův B-lymfom centrálního nervového systému
- Neuropsychiatrické symptomy jako časná manifestace Alzheimerovy nemoci