
Dva případy atypického teratoidního rhabdoidního tumoru CNS a přehled literatury
Two cases of an atypical teratoid rhabdoid tumour of the CNS and literature review
Atypical teratoid rhabdoid tumour is a highly malignant tumor of central nervous system predominantly affecting children under 3 years of age. It is a rare yet severe condition.
Diagnostics of this entity might be problematic due to unspecific histological and imiging findings which may mimic characteristics of other pediatric brain tumors. Therefore, exact methods of immunohistochemistry and molecular biology are required for the final diagnosis.
Historically, this condition has been associated with poor prognosis. Fortunately, recent studies have brought more optimistic results, specifically with the use of intensive multimodal therapy containing a combination of surgical resection, chemotherapy including intrathecal administration and focal radiotherapy.
The authors present two cases recently treated at our department as well as a review of scientific literature concerning this issue.
Key words:
atypical teratoid rhabdoid tumor – pediatric embryonal tumor – central nervous systém – primary resection – molecular a immunohistochemical diagnostics – multimodal adjuvant therapy
Autoři:
H. Zítek 1; D. Sumerauer 2; J. Zámečník 3; P. Vachata 1; M. Sameš 1; A. Puchmajerová 4
Působiště autorů:
Neurochirugická klinika UJEP a Masarykovy nemocnice Ústí nad Labem
1; Klinika dětské hematologie a onkologie 2. LF UK a FN Motol
2; Ústav patologie a molekulární medicíny 2. LF UK a FN Motol
3; Ústav biologie a lékařské genetiky 2. LF UK a FN Motol
4
Vyšlo v časopise:
Cesk Slov Neurol N 2017; 80/113(Online only): 0
Kategorie:
č. 5
Souhrn
Atypický teratoidní rhabdoidní tumor patří mezi vysoce maligní nádory centrální nervové soustavy postihující nejčastěji děti do 3 let věku. Jde o vzácnou, nicméně závažnou diagnózu.
Problémem u této jednotky může být již samotná diagnostika, jelikož histologické a grafické nálezy mohou imitovat jiné častější nádory centrální nervové soustavy. K diagnóze je tedy vyžadován důkaz inaktivace genu SMARCB1/INI1, popřípadě genu SMARCA4 pomocí imunohistochemických či molekulárně genetických metod. Toto onemocnění bylo historicky spojeno s infaustní prognózou. V posledních letech se nicméně objevily práce, které přinesly optimističtější výsledky díky intenzivní multimodální terapii, tedy kombinaci resekce, vysokodávkované chemoterapie, včetně chemoterapie intratékální, a fokální radioterapie.
Autoři prezentují dva případy, které se v nedávné době vyskytly na našem pracovišti, a přehled odborné literatury věnující se této problematice.
Klíčová slova:
atypický teratoidní rhabdoidní tumor – embryonální tumor – centrální nervová soustava – primární resekce – molekulárně-biologická a imunohistochemická diagnostika – multimodální adjuvantní terapie
Úvod
Atypický teratoidní rhabdoidní tumor (AT/RT) je vzácný maligní (WHO grade IV) embryonální nádor centrální nervové soustavy (CNS) dětského věku. Představuje méně než 5 % všech dětských tumorů CNS. Podíl roste u nejmenších dětí a nejčastější je u dětí do 3 let věku, kde představuje až 20 % tumorů CNS [1–6]. Jde o nádor velmi agresivní, který se projevuje progresivní a vážnou symptomatikou a vede zpravidla k infaustnímu konci. Buscariollo et al publikovali soubor 144 pacientů, kde průměrné celkové přežití bylo 10 měsíců [4]. Práce z posledních let však přinášejí optimističtější závěry týkající se celkového přežití těchto pacientů, a to díky intenzivní multimodální terapii [4, 7–9].
AT/RT je relativně nová jednotka, která byla poprvé popsána v roce 1987 [10], a na seznam Světové zdravotnické organizace se definitivně dostala až v roce 2000 [11]. Zpočátku bylo AT/RT často zaměňováno za jiné tumory CNS typické pro dětský věk, především meduloblastom, primitivní neuroektodermální tumor či karcinom choroidálního plexu. K tomu vedla především histologická stavba tkáně tohoto nádoru, jelikož ta, kromě typických rhabdoidních buněk, může obsahovat také buňky mezenchymální, epiteliální či buňky neuroektodermální [12]. Definitivní možnost rozlišení tohoto nádoru od výše zmíněných jednotek přinesly molekulárně biologické a imunohistochemické metody. Bylo zjištěno, že většina těchto nádorů je charakterizována delecí či inaktivační mutací tumor supresorového genu SMARCB1/INI1/hSNF5 na chromozomu 22q1, a to germinální či získanou [13]. Ztráta nukleární exprese tohoto genu při imunohistochemickém vyšetření je pro tuto jednotku patognomická. Primární lokalizace AT/RT může být kdekoliv v rámci CNS, nicméně převažují intrakraniální lokalizace. Nádory tohoto typu jsou schopny vytvářet metastázy v rámci CNS, které dle některých autorů dále zhoršují prognózu tohoto onemocnění [4, 14, 15]. Práce, ve kterých pacienti s metastatickým postižením podstoupili multimodální terapii, však vliv metastáz na prognózu nepopisují [7, 14, 16].
Dle našich zjištění se tomuto nádoru v české neurochirurgické literatuře dosud nevěnovala přílišná pozornost. Prezentujeme tedy 2 kazuistiky z našeho pracoviště, které mají mimo jiné upozornit odbornou společnost, aby zařadila tuto jednotku do diferenciálně-diagnostického uvažování v rámci neuroonkologie dětských pacientů.
Kazuistika č. 1
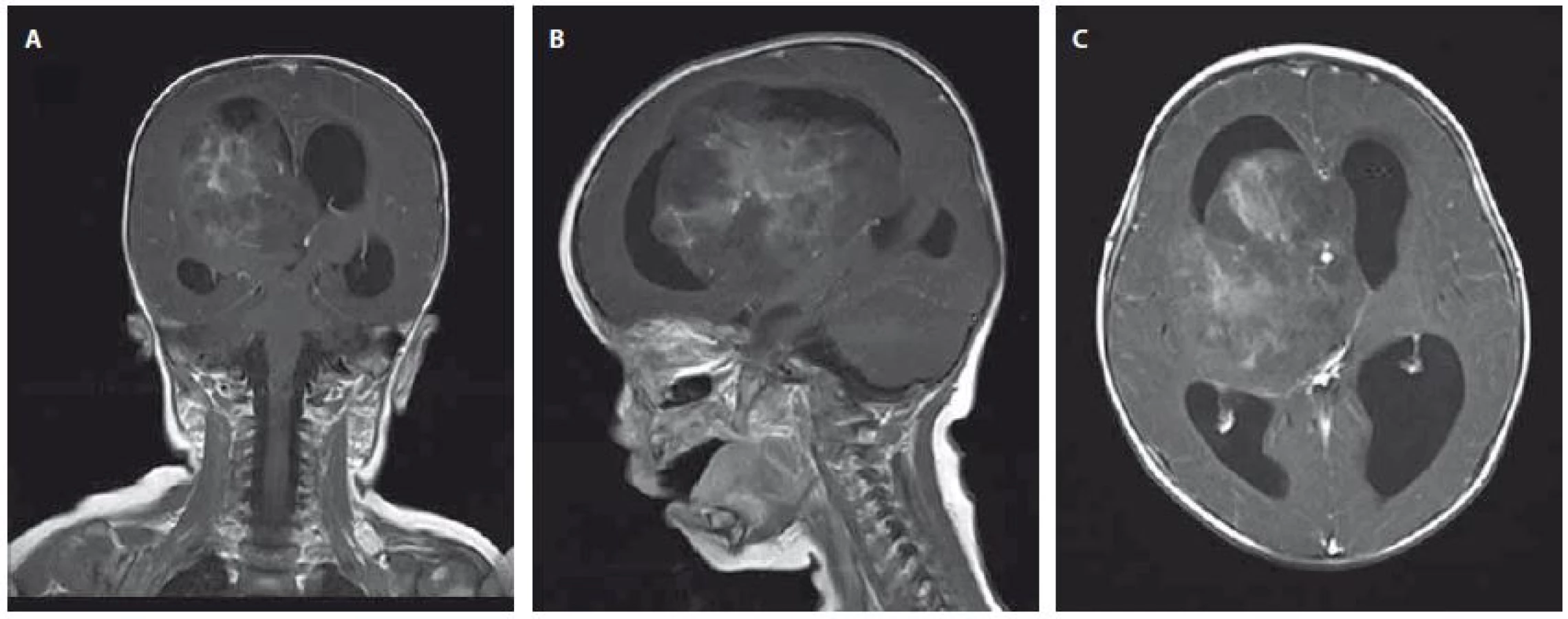
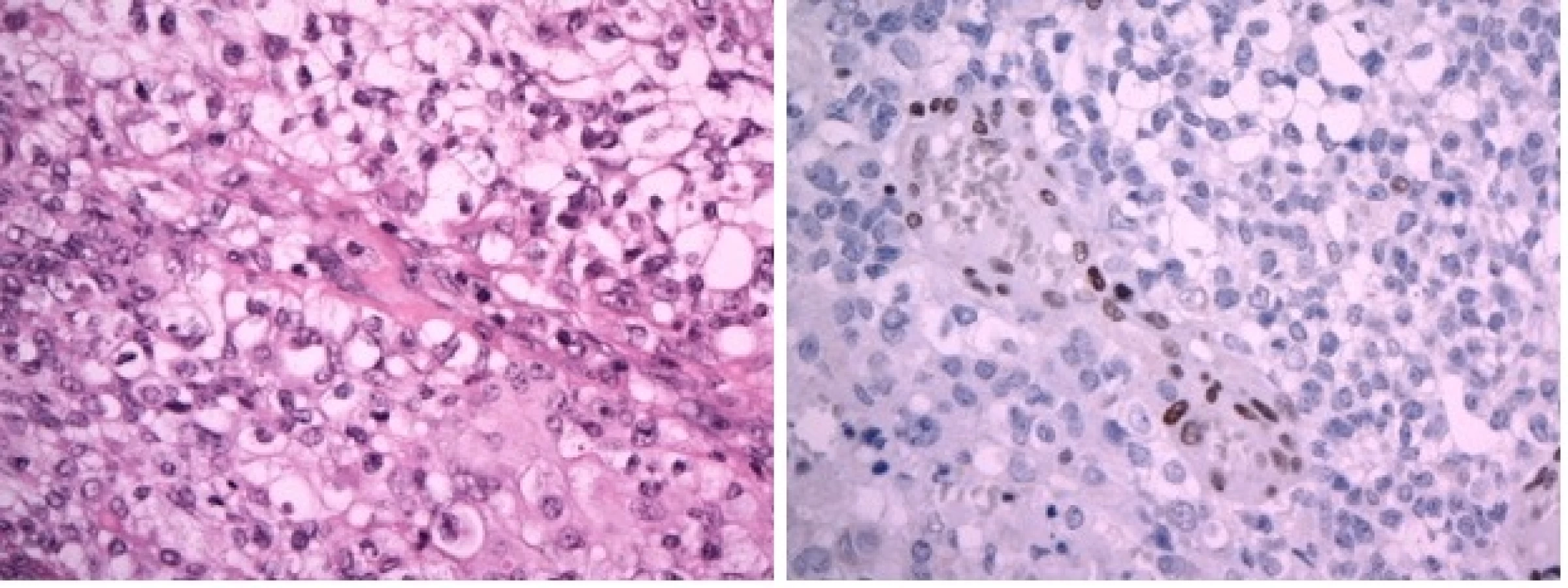
Devítiměsíční kojenec byl přijat na JIP dětské kliniky pro 3 dny trvající levostrannou hemiparézu, plačtivost a neklid. Dle matky měl také za poslední dva dny menší chuť k jídlu. Při příjmu byl dále zjištěn obvod hlavičky nad 97. percentilem a regres v psychomotorickém vývoji. Na základě klinického obrazu bylo doplněno MR mozku, kde byla popsána velká expanze 56×91×76 mm zasahující do III. a pravé postranní mozkové komory se známkami akutního hydrocefalu a přesunem středočárových struktur o 16 mm doleva. Po podání kontrastní látky se tumor nehomogenně sytí (obr. 1A–1C). Během 24 hod klinický stav dále progredoval. Pacient prodělal sekundární epileptický záchvat s křečemi na pravostranných končetinách a následně byl popsán obraz centrální tentoriální herniace s bilaterální mydriázou. Pacient byl následně zaintubován a byl indikován urgentní operační výkon z vitální indikace. Perioperační obraz byl dramatický v podobě vysokého tlaku mozkomíšního moku a významného krvácení z léze. Byla provedena subtotální resekce a bazální stomie III. komory s úspěšnou stázou excesivního krvácení. Na kontrolním CT však byl popsán obraz difúzního edému mozku se závažnou prognózou a bez indikace k další neurochirurgické intervenci. Desátý den od prvních příznaků byla definitivně stanovena smrt mozku a pacient své nemoci podlehl. Pozdější výsledek histologie potvrdil klasifikaci tumoru jako AT/RT s imunohistochemickou negativitou INI-1 včetně pozitivní mutační analýzy genu SMARCB1/INI1 (obr. 2A, 2B).

Kazuistika č. 2
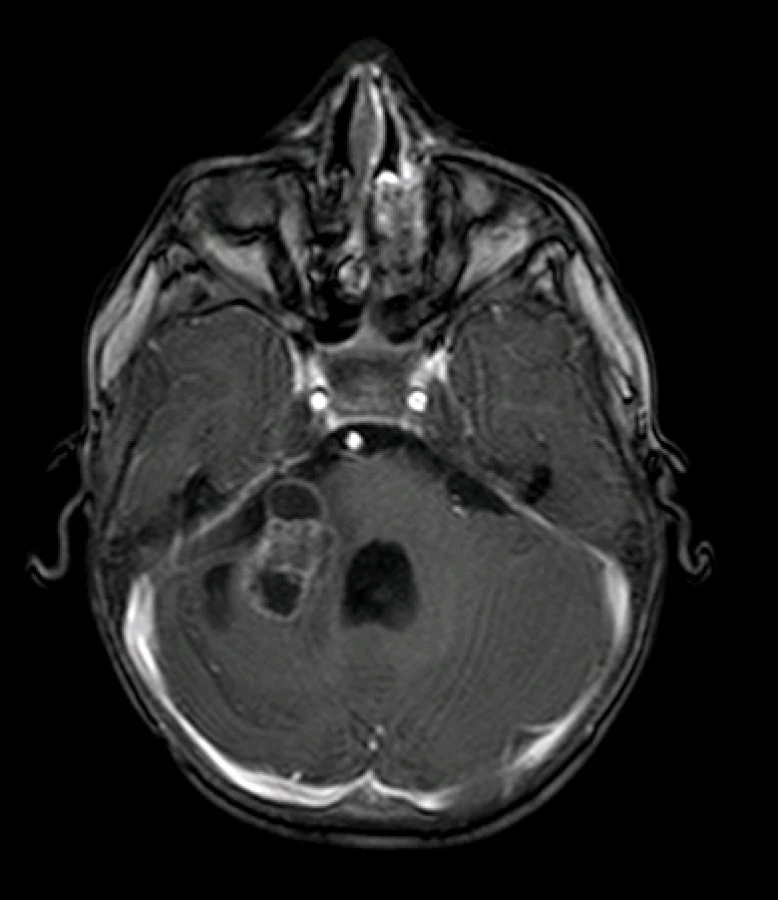
Pacient starý 23 měsíců byl hospitalizován pro asi půl roku trvající neprospívání, intoleranci p.o. příjmu a opakované zvracení. V průběhu měsíce je pro uvedené obtíže opakovaně hospitalizován na spádové pediatrii k enterální a parenterální rehydrataci, po které se stav dítěte přechodně zlepší, ale po 2–3 dnech se zvracení a intolerance p.o. příjmu opakuje. Bylo doplněno neurologické konzilium s nálezem centrálního hypotonického syndromu, vestibulocerebelárního syndromu, pravostranné periferní parézy n. VII, horizontálního nystagmu a parézy n. VI vpravo. Na vyšším pracovišti je provedena MR mozku s nálezem nehomogenně se sytícího ložiska v zadní jámě lební vpravo o velikosti 45×32×41 mm infiltrující pravou mozečkovou hemisféru a kmen a další podobná ložiska v zadní jámě vlevo, v oblasti ganglion Gasseri vlevo a pravém hippokampu. Komorový systém je supratentoriálně rozšířený se známkami incipientní dekompenzace hydrocefalu (obr 3A–3C).
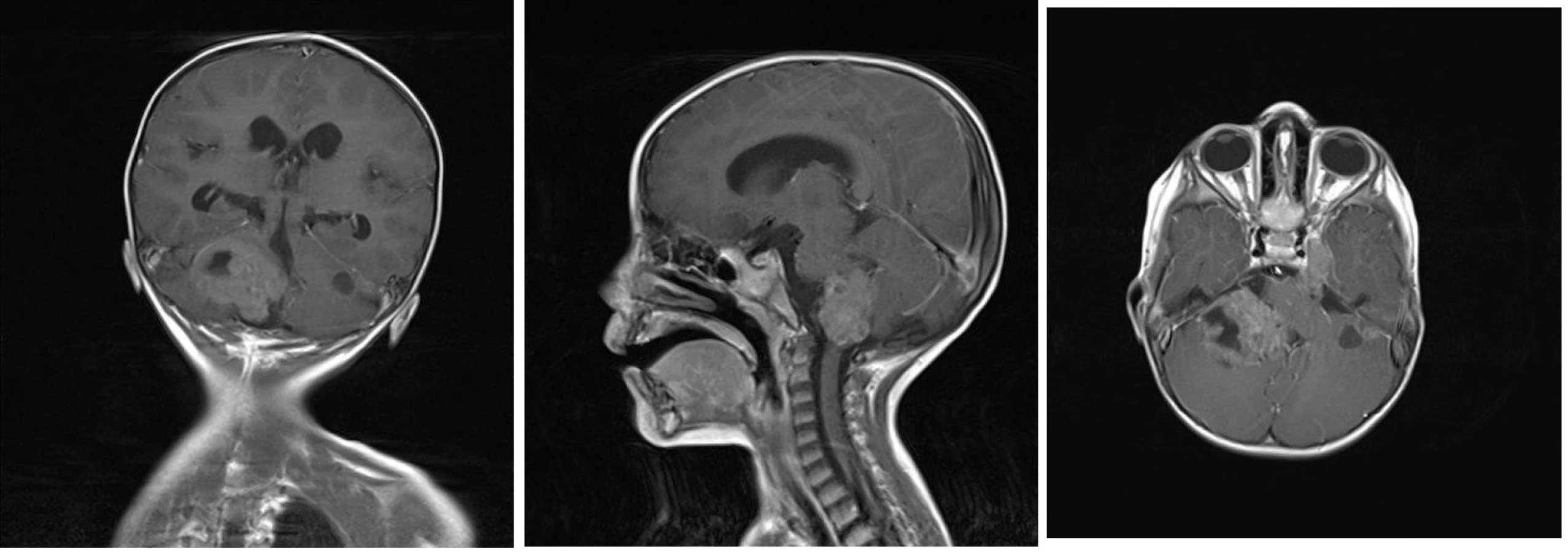
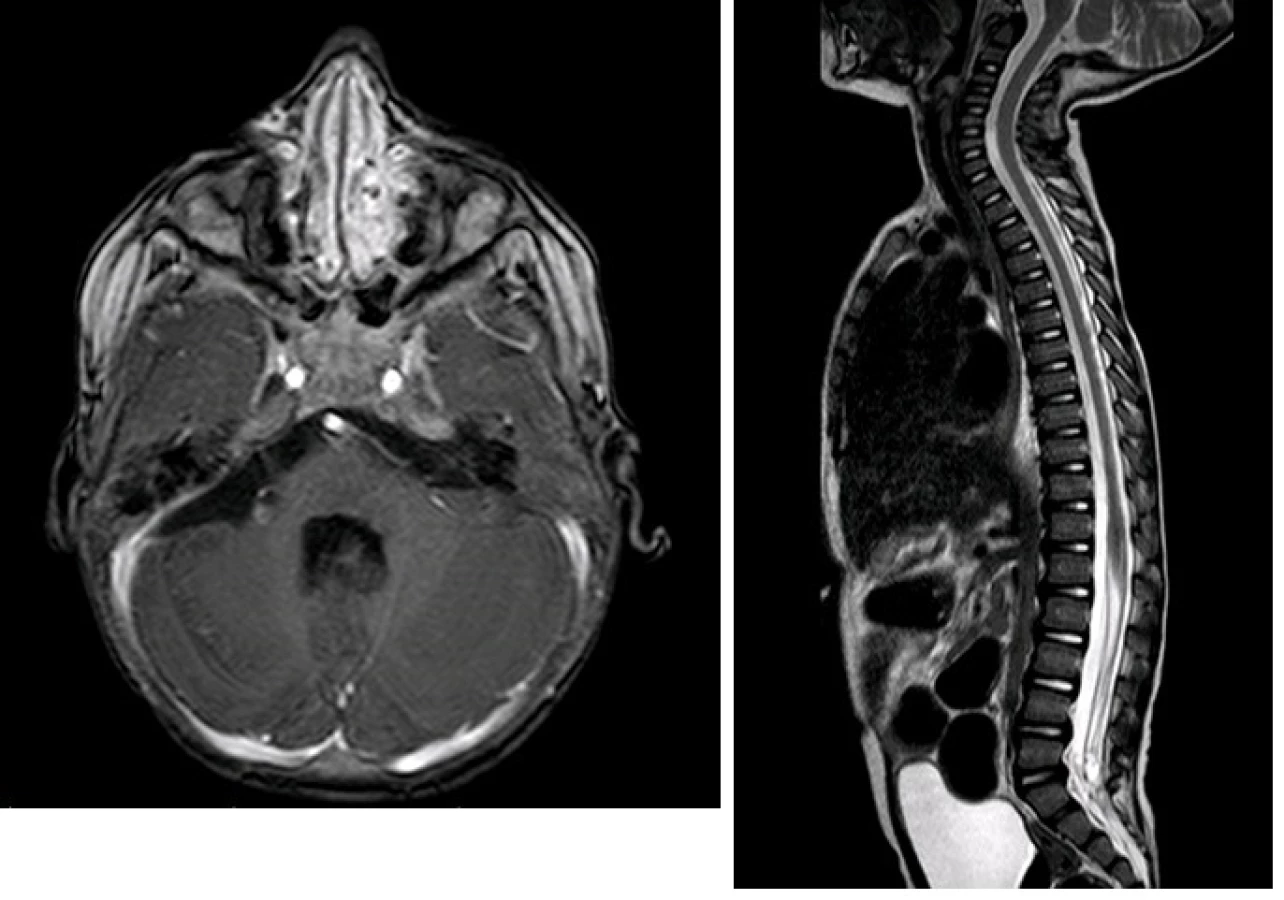
Nejprve jsme přistoupili k operačnímu řešení. Byla provedena parciální resekce tumoru z retrosigmoidálního přístupu vpravo. Histologicky je zastižen hypercelulární nádor pestré stavby s ložisky nekrotizace. Nádor převážně sestává ze solidně alveolárně uspořádaných drobných polygonálních buněk se světlou nebo jemně eozinofilní cytoplazmou. Diagnosticky cenným vyšetřením je průkaz jaderné negativity proteinu INI–1 v nádoru imunohistochemickým vyšetřením. Morfologický obraz a imunohistochemické vyšetření i prokázaná mutace v genu SMARCB1/INI1 vedly k diagnóze vysoce maligního embryonálního nádoru mozku dětského věku – atypický teratoidní rhabdoidní tumor (AT/RT), WHO grade IV (obr. 4A, 4B). V pooperačním období u chlapce dominuje mutizmus, povšechná hypotonie, bulbární symptomatologie, pooperační průběh je komplikován aspirační pneumonií při poruše polykání a přechodnou sekrecí likvoru z operační rány. K další léčbě je chlapec přeložen na Kliniku dětské hematologie a onkologie FN v Motole. Za intenzivní rehabilitace byla doplněna stagingová vyšetření. MR mozku a míchy potvrzující vícečetná ložiska AT/RT supra- a infratentoriálně a navíc další metastatická ložiska v oblasti páteřního kanálu, největší v oblasti Th11–L1 a L4–S2 (obr. 5). V mozkomíšním moku odebraném z lumbální punkce jsou prokázány 4 % atypických nádorových elementů. Vzhledem k diagnóze AT/RT, možnosti synchronních rhabdoidních nádorů ledvin a měkkých tkání je vstupně provedena celotělová MR k vyloučení současného extrakraniálního postižení. Pro závažný stav dítěte, neurologický deficit je bezprostředně zahájena chemoterapie podle doporučení EU-RHAB protokolu kombinující systémové a intratékální podání cytostatik. Postupně se zlepšuje hybnost, chlapec je vertikalizován, schopen sedu a chůze s dopomocí. Zlepšuje se bulbární symptomatologie. Kontrolní MR mozku a míchy prokazuje postupnou významnou regresi nádoru ve všech lokalizacích, atypické elementy v mozkomíšním moku již neprokazovány. Po devíti blocích chemoterapie chlapec ve velmi dobré parciální remisi podstoupil vysokodávkovanou chemoterapii režimem karboplatina/thiotepa s podporou autologních kmenových hematopoetických buněk. Vzhledem k věku a vysokému riziku recidivy onemocnění v potransplantačním období pokračuje chlapec v protinádorové léčbě metronomickým režimem kombinujícím nízcedávkovanou p.o. chemoterapii a intratékalní aplikace lipozomálního cytosinarabinosidu. Radioterapie s ohledem na věk dítěte a rozsah ozařovacího pole nebyla v primární léčbě indikována.

Výsledkem této kombinované terapie byla stabilizace onemocnění s přechodnou remisí dle grafického nálezu (obr. 6A, 6B), 13 měsíců od primární operace však dochází k progresi klinického obrazu, zejména zhoršení bulbárního syndromu a kvadruparézy. Na kontrolní MR je prokázána recidiva tumoru v mostomozečkovém koutu vpravo částečně infiltrující mozkový kmen (obr.7). Vzhledem ke klinickému obrazu a infaustní prognóze při recidivě bylo po domluvě s rodiči pacienta rozhodnuto o ukončení onkologické terapie. Pacient v následujících dnech svému onemocnění podléhá.
Diskuze
AT/RT tvoří méně než 5 % dětských tumorů CNS, ale až 20 % tumorů CNS u dětí do 3 let věku. Pravá incidence však může být vyšší vzhledem k histologické a radiodiagnostické podobnosti s jinými maligními nádory CNS u dětí. V dospělém věku jde o extrémně vzácný nádor a byly popsány jen jednotlivé případy [1–6]. Diagnostika tohoto onemocnění se kromě klinického obrazu opírá především o radiodiagnostiku, histologický, molekulární a imunohistochemický rozbor bioptické tkáně a cytologický rozbor mozkomíšního moku.
AT/RT postihuje typicky nejmenší děti, jejichž stav se často progresivně, během dnů či týdnů, zhoršuje. Dominující symptomatika se odvíjí především od lokalizace a rychlosti růstu ložiska. Často se vyskytují příznaky hydrocefalu, tedy bolesti hlavy, zvracení, letargie, narušení psychomotorického vývoje a v neposlední řadě růst obvodu hlavy. Výjimkou nejsou ani náhle vzniklé hemiparézy, ataxie, sekundární epileptické záchvaty či obrny hlavových nervů. [17–19] Progresivní neurotopická symptomatika byla přítomna v obou námi prezentovaných případech.
Atypický teratoidní rhabdoidní tumor se vyskytuje ve všech částech CNS bez výrazné predilekce mezi infra- a supratentoriální lokalizací [4,5,14,16,20]. Primární postižení míchy bývá přítomno v 4 % [16,19]. Na CT zobrazení má solidní složka této léze isodenzní či lehce hyperdenzní obraz s hypodenzními okrsky cystické a nekrotické složky. Především u nádorů zasahujících do zadní jámy lební nepřekvapí grafické známky hydrocefalu, které byly patrné u obou našich pacientů. Mohou být také zjevné intralezionální kalcifikace. Na MR se tumor zobrazuje jako heterogenní léze, která nemá typický obraz na T1 či T2 vážených obrazech, a stejně tak se ložiska mohou, ale také nemusí, sytit při podání gadolinia [21]. Na DWI sekvenci nese tento tumor známky restrikce difúze [22]. Pacienti s podezřením na toto onemocnění by také měli podstoupit vyšetření celého CNS, vzhledem k možnému výskytu metastáz. Jejich četnost se dle dostupné literatury pohybuje kolem 30 % [7,16,20] a dle některých autorů jsou častější u dětí mladších než 3 roky [2]. Vícečetné metastatické postižení bylo diagnostikováno i v jedné z našich kazuistik.
Jak již bylo uvedeno, histologický obraz bioptické tkáně může být často velmi variabilní a histogeneze tohoto onemocnění není doposud zjištěna. V různém zastoupení bývají popisovány buňky charakteru primitivních neuroektodermálních, mezenchymálních, epiteliálních a typické rhabdoidní buňky. Poslední zmíněné jsou pro tento tumor patognomické, a jsou tedy cílem histologického rozboru. Jde o velké buňky s bohatou eozinofilní cytoplazmou s inkluzemi tvořenými nakupením intermediárních filament, excentricky uloženým jádrem s patrným jadérkem. Zastoupení těchto buněk je však často ve vzorcích velmi variabilní a v některých případech nejsou zastiženy vůbec, což dále ztěžuje diagnostiku [9,12]. K diagnóze naopak přispívá nutné imunohistochemické vyšetření, které prakticky ve všech případech, včetně našich dvou kazuistik, prokáže absenci nukleární exprese INI–1 [2,14,19,20]. Tento marker je nejužitečnější v rámci diferenciální diagnostiky maligních tumorů CNS a byl pozitivní v obou námi popisovaných případech. Molekulární vyšetření dále prokáže mutaci či deleci v genu SMARCB1/INI–1/hSNF5 na chromozomu 22q1 či vzácně alterace v genu SMARCA4. Mutace však nemusí být geneticky vždy prokázána, a imunohistochemické vyšetření je tedy považováno za sensitivnější.
Malé procento AT/RT má však expresi INI–1 zachovalou, a zkoumání dalších markerů je tudíž zásadní [20].
Prognóza pacientů s tímto onemocněním byla historicky špatná. Přispívala k tomu nejen vysoká agresivita těchto malignit, ale také dlouhou dobu chybějící efektivní léčebný protokol. Vzhledem k tomu, že jednotka AT/RT byla popsána relativně recentně a jde o vzácnou jednotku, léčebné standardy stále chybí. V poslední době však byly dosaženy optimistické výsledky agresivní multimodální terapií v podobě chirurgie, intenzivní chemoterapie a radioterapie. S ohledem ke vzácnosti onemocnění a náročnosti komplexní terapie by tato měla probíhat ve specializovaných onkologických centrech disponujících kompletním terapeutickým zázemím [30]. Prvním terapeutickým krokem bývá chirurgická resekce tumoru. V literatuře nejsou jednoznačné závěry, zda rozsah a radikalita operačního zákroku zlepšuje prognózu v podobě celkového přežití či přežití bez relapsu. Hilden et al [1], Chi et al [7] a Lafay-Cousin et al [16] popsali, že děti, které podstoupily radikální resekci, mají lepší prognózu než ty, které podstoupily jen subtotální resekci či biopsii. Nicméně ve větších souborech pacientů již není rozdíl v době přežití u pacientů léčených chemoterapií dle radikality operace signifikantní. Tyto tumory také často dosahují velkých rozměrů a nacházejí se v chirurgicky obtížně dostupných či rizikových oblastech CNS. Z tohoto pohledu se tedy zdá, že dosažení radikální resekce i za cenu rizika komplikací není u těchto lézí na místě a ani v našich dvou případech nebyla resekce tumoru radikální [6,14].
Rozhodující adjuvantní terapeutickou modalitou se ukazuje být chemoterapie. Poslední léta byla právě ve znamení zkoušení dosavadních chemoterapeutických protokolů a jejich přizpůsobování tomuto onemocnění. Velká variabilita použitých protokolů, jejich kombinace s radioterapií (s nebo bez), ale i různé dávkování těchto chemoterapeutik a načasování jejich podání do značné míry znemožňují porovnání jednotlivých režimů z pohledu jejich vlivu na celkové přežití. První články popisovaly špatnou prognózu u pacientů léčených konvenční CHT [23,24]. Výsledky později publikovaných studií se přiklonily k vysokodávkované chemoterapii nebo také kombinaci systémové a intratékální chemoterapie. Vysokodávkovaná chemoterapie byla vzhledem ke své významné toxicitě doplněna o autologní transplantaci hematopoetických kmenových buněk. Tato kombinace byla spojena s lepší prognózou, ale počet zdokumentovaných případů zůstává malý [6]. Optimističtějších výsledků bylo také dosaženo s použitím režimu na základě protokolu pro terapii rhabdomyosarkomu s parameningeální lokalizací z Intergroup Rhabdomyosarcoma Study–III (IRS–III). Chi et al [7] popsali soubor 20 pacientů, u nichž byla použita kombinace IRS–III, intratékální terapie a radioterapie (konformační či ozáření kraniospinální osy). Dvouleté celkové přežití a přežití bez relapsu nemoci činilo 53 a 70 %, a tedy kombinace IRS–III a radioterapie může být efektivním postupem při léčbě AT/RT. Slavc et al [9] publikovali soubor 22 pacientů, z nichž 13 (kohorta A) léčených intenzivním multimodálním režimem zahrnujícím doxorubucin, cyklofosfamid, vinkristin, ifofosfamid, cisplatinu, etoposid a metotrexát, doplněným o opakované intratékální podání a následovaným vysokodávkovanou chemoterapií s autologní transplantací hematopoetických kmenových buněk. Léčba byla na závěr doplněna o lokální radioterapii. Kohorta B byla původně léčena pro jinou diagnózu jinými léčebnými protokoly podle typu původního tumoru, než byla diagnóza retrospektivně upravena. Pětileté celkové přežití a přežití bez relapsu bylo v kohortě B 28,8 % a v kohortě A pak 100 % resp. 88,9 %. V kohortě A tedy nebylo pozorováno úmrtí při mediánu délky přežití 76 měsíců. Intratékální chemoterapie (IT) je v rámci multimodálního protokolu spojována s delším přežitím, především vzhledem k jejímu předpokládanému vlivu na vzdálený relaps [7,14,16]. Náš druhý pacient taktéž podstoupil vysokodávkovanou chemoterapii včetně opakovaného intratékálního podání metotrexátu a doplněnou o transplantaci hematopoetických kmenových buněk. V kombinaci s primární resekcí došlo k dočasné stabilizaci onemocnění s parciální regresí ložisek na MR.
Na rozdíl od chemoterapie, která je nedílnou součástí léčby tohoto onemocnění, je užití radioterapie (RT) kontroverznější. Jak jsme se již zmínili, toto onemocnění postihuje nejmladší pacienty, u kterých jsou dobře popsány nežádoucí účinky užití radioterapie v podobě narušení vývoje CNS a z toho plynoucí dlouhodobé komplikace. Z tohoto důvodu byla RT u nejmladších pacientů odkládána či indikována jen k terapii recidiv. V poslední době však autoři poukazují na významnou souvislost mezi dobou přežití a užití RT v první linii adjuvantní terapie [3,14,15,26]. Vzhledem k rizikovosti pozdních následků při použití RT u malých dětí, mělo by vždy jít o vysoce fokální ozáření a difúzní ozáření ponechat pro starší pacienty, u kterých by mohla vést k lepší kontrole diseminace a relapsu onemocnění [26]. Slavc et al [9] dále navrhují doplnit intratékální chemoterapii v případě, že pacient podstoupí pouze fokální ozáření jako prevenci leptomeningeální diseminace. Podobný závěr prezentovali i Hirth et al při užití kombinace IT a Leksellova gama nože [27]. Objevují se ale i opačné názory. Lafay et al [16] neprokázali efektivitu RT na celkové přežití a 6 z 11 jejich dlouhodobě přežívajících pacientů podstoupilo pouze vysokodávkovanou chemoterapii s transplantací autologních kmenových buněk bez jakéhokoliv ozáření.
Předpokládá se, že přibližně jedna třetina pacientů s nově diagnostikovaným AT/RT jsou nositeli germinální mutace genu SMARCB1 nebo vzácně SMARCA4. Mluvíme poté o tzv. Rhabdoid Tumor Predisposition Syndrome. Na podkladě tohoto zjištění by rodinám pacientů měla být nabídnuta genetická konzultace [28,29].
V roce 2005 se začal formovat Evropský rhabdoidní registr (EU-RHAB), jehož cílem je podávat výše zmíněné odborné informace týkající se diagnostických a terapeutických postupů u rhabdoidních tumorů včetně AT/RT.
Závěr
Atypický teratoidní rhabdoidní tumor patří mezi vzácná a smrtící onemocnění. Na základě podobnosti s ostatními dětskými maligními nádory CNS se lze domnívat, že jde poddiagnostikovanou jednotku. Odlišení od ostatním podobných jednotek je ale zásadní pro odlišnosti v prognóze a terapii tohoto tumoru. V poslední době se množí práce, které publikují optimističtější výsledky v léčbě, a to především zásluhou multimodální terapie. Tato intenzivní léčba se však vyznačuje nezanedbatelnou toxicitou, a tedy – až bude standardizována terapie zajišťující podstatné zlepšení prognózy těchto pacientů – bude v budoucnu nutné řešit prevenci vzniku neurokognitivních, endokrinologických komplikací a sekundárních malignit v souvislosti s touto terapií.
MUDr. Hynek Zítek
Neurochirugická klinika UJEP a Masarykovy nemocnice Ústí nad Labem
Sociální péče 3316/12A
401 13 Ústí nad Labem
email: hynek.zitek@kzcr.eu
Přijato k recenzi: 4. 7. 2017
Přijato do tisku: 3. 8. 2017
Zdroje
1. Buscariollo DL, Park HS, Roberts KB, et al. Survival outcomes in atypical teratoid rhabdoid tumor for patients undergoing radiotherapy in a Surveillance, Epidemiology, and End Results analysis. Cancer 2012;118(17):4212–19. doi: 10.1002/cncr.27373.
2. Schrey D, Carceller Lechón F, Malietzis G, et al. Multimodal therapy in children and adolescents with newly diagnosed atypical teratoid rhabdoid tumor: individual pooled data analysis and review of the literature. J Neurooncol 2016;126(1):81–90. doi: 10.1007/s11060-015-1904-0.
3. Slavc I, Chocholous M, Leiss U, et al. Atypical teratoid rhabdoid tumor: improved long-term survival with an intensive multimodal therapy and delayed radiotherapy. The Medical University of Vienna Experience 1992–2012. Cancer Med 2014;3(1):91–100. doi: 10.1002/cam4.161.
4. DiPatri AJ, Sredni ST, Grahovac G, et al. Atypical teratoid rhabdoid tumors of the posterior fossa in children. Childs Nerv Syst 2015;31(10):1717–28. doi: 10.1007/s00381-015-2844-x.
5. Ostrom QT, Chen Y, M de Blank P, et al. The descriptive epidemiology of atypical teratoid/rhabdoid tumors in the United States, 2001-2010. Neuro Oncol 2014;16(10):1392–9. doi: 10.1093/neuonc/nou090.
6. Lafay-Cousin L, Hawkins C, Carret AS, et al. Central nervous system atypical teratoid rhabdoid tumours: the Canadian Paediatric Brain Tumour Consortium experience. Eur J Cancer 2012;48(3):353–9. doi: 10.1016/j.ejca.2011.09.005.
7. Dufour C, Beaugrand A, Le Deley MC, et al. Clinicopathologic prognostic factors in childhood atypical teratoid and rhabdoid tumor of the central nervous system: a multicenter study. Cancer 2012;118(15):3812–21. doi: 10.1002/cncr.26684.
8. Chi SN, Zimmerman MA, Yao X, et al. Intensive multimodality treatment for children with newly diagnosed CNS atypical teratoid rhabdoid tumor. J Clin Oncol 2009;27(3):385–9. doi: 10.1200/JCO.2008.18.7724.
9. von Hoff K, Hinkes B, Dannenmann-Stern E, et al. Frequency, risk-factors and survival of children with atypical teratoid rhabdoid tumors (AT/RT) of the CNS diagnosed between 1988 and 2004, and registered to the German HIT database. Pediatr Blood Cancer 2011 Dec 1;57(6):978–85. doi: 10.1002/pbc.23236.
10. Pai Panandiker AS, Merchant TE, Beltran C, et al. Sequencing of local therapy affects the pattern of treatment failure and survival in children with atypical teratoid rhabdoid tumors of the central nervous system. Int J Radiat Oncol Biol Phys 2012;82(5):1756–63. doi: 10.1016/j.ijrobp.2011.02.059.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie
2017 Číslo Online only
Nejčtenější v tomto čísle
- Dva případy atypického teratoidního rhabdoidního tumoru CNS a přehled literatury
- Intravenózní trombolýza po zrušení účinku dabigatranu specifickým antidotem Idarucizumabem
- Kazuistika meningitidy dospělých způsobená bakterií Escherichia coli