
Role specifické buněčné imunity v patogenezi roztroušené sklerózy se zaměřením na Th17 a Treg lymfocyty
The Role of the Cell-mediated Immunity in the Pathogenesis of Multiple Sclerosis with Focus on Th17 and Treg Lymfocytes
Multiple sclerosis is a serious autoimmune disease of the central nervous system. It occurs with relatively high prevalence, especially in young people. It is essential to understand the pathogenesis of this disease in order to develop new treatments. All components of immunity are involved in this process but current research mainly focuses on lymphocyte populations. Previously, imbalance between subtypes of helper lymphocytes Th1 and Th2 was considered as the main cause of multiple sclerosis. Recently, the influence of other cell elements, such as B lymphocytes, cytotoxic T lymphocytes, was shown. Moreover, new cell types, regulatory T lymphocytes and helper Th17 lymphocytes, have been discovered. The aim of this article is to describe the main roles of individual lymphocyte subtypes in multiple sclerosis pathogenesis, focusing first on regulatory T lymphocytes and helper Th17 lymphocytes.
Key words:
multiple sclerosis – B lymphocytes – T lymphocytes – regulatory T lymphocytes – Th17 lymphocytes
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Autoři:
M. Svobodová 1; P. Štourač 1,2
Působiště autorů:
Neurologická klinika LF MU a FN Brno
1; CEITEC – Středoevropský technologický institut, MU, Brno
2
Vyšlo v časopise:
Cesk Slov Neurol N 2017; 80/113(2): 173-179
Kategorie:
Přehledný referát
doi:
https://doi.org/10.14735/amcsnn2017173
Podpořeno grantem MZ ČR – RVO (FNBr, 65269705) a projektem specifického výzkumu č. MUNI/ A/ 1072/ 2015 z programu podpory studentských projektů na Masarykově univerzitě.
Souhrn
Roztroušená skleróza je závažné autoimunitní onemocnění centrální nervové soustavy. Vyskytuje se s relativně vysokou prevalencí, a to především u mladých lidí. K vývoji nových léčiv je zásadní pochopení patogeneze tohoto onemocnění. Té se účastní všechny složky imunity. Zkoumány jsou především lymfocytární populace. Dříve se za hlavní příčinu roztroušené sklerózy považovala nerovnováha mezi subtypy helperských lymfocytů Th1 a Th2. V posledních letech byl ale prokázán vliv dalších buněčných elementů, jako jsou B lymfocyty, cytotoxické T lymfocyty, a byly objeveny i nové buněčné typy, regulační T lymfocyty a helperské Th17. Tento článek má za cíl přiblížit základní role jednotlivých lymfocytárních podtypů v rozvoji roztroušené sklerózy, a to se zaměřením především na regulační T lymfocyty a pomocné Th17.
Klíčová slova:
roztroušená skleróza – B lymfocyty – T lymfocyty – regulační T lymfocyty – Th17 lymfocyty
Úvod
Roztroušená skleróza (RS) je autoimunitní onemocnění postihující centrální nervovou soustavu (CNS). Jde o onemocnění s častým výskytem, v České republice s prevalencí 100– 150/ 100 000 [1]. Objev nových léčebných možností zpomalujících progresi nemoci závisí především na pochopení patogeneze. Na vývoji RS se podílejí všechny složky imunitního systému (imunita nespecifická i specifická, humorální i buněčná). K současnému poznání významně přispěly studie na animálních modelech pomocí tzv. experimentální autoimunitní encefalomyelitidy (EAE) [2].
U pacientů s RS jsou v CNS přítomna mnohočetná perivaskulární zánětlivá ložiska (plaky). V nich nacházíme CD4+ a CD8+ T lymfocyty, aktivované makrofágy, mikroglie a B lymfocyty. Ve většině infiltrátů také protilátky a složky komplementu [3].
Dlouho byla za základní imunopatogenetickou podstatu RS považována především porušená rovnováha mezi Th1 a Th2. Th1 jsou považovány za „induktory“ zánětu, naopak Th2 za protizánětlivé. Při poruše rovnováhy diferenciace Th1/ Th2 dochází k patologiím. Při převaze Th1 jsou to zánětlivá a autoimunitní onemocnění, při převaze Th2 atopické a nádorové stavy [4].
Směr diferenciace prekurzorové CD4+ buňky v Th1/ Th2 je dán poměrem koncentrací cytokinů IL-12 a IL-4. IL-12 je produkován makrofágy a dendritickými buňkami a stimuluje vznik Th1. IL-4 je produkován hlavně bazofily a mastocyty, stimuluje vznik Th2 [5,6].
Ukázalo se ale, že tato představa byla velmi zjednodušená. V roce 1995 Sakaguchi et al objevili nový podtyp T lymfocytů, regulační (Treg). Když k tomu v roce 2005 přibyl objev nových helperských T buněk, Th17, bylo jasné, že situace je mnohem složitější. Stejně jako Th1 mají i Th17 prozánětlivou roli. Fyziologicky proti jejich funkci působí Treg, které zabraňují poškození tkání jejich nadměrnou aktivitou. Při vzniku autoreaktivních klonů Th17 může dojít ke vzniku autoimunitního onemocnění. To se vyvíjí pravděpodobně při početní nebo funkční nedostatečnosti Treg [7,8].
Do patogeneze RS zasahují také B lymfocyty. Roli hraje jejich protilátková produkce, ale i interakce s T lymfocyty, produkce cytokinů a také jejich regulační aktivita.
Specifická imunita buněčná
Tyto buňky patří do lymfoidní linie. Téměř celý vývoj B lymfocytů probíhá v kostní dřeni a dokončuje se v sekundárních lymfoidních orgánech po setkání s antigenem. Diferencují se v plazmatické buňky produkující protilátky. Naopak hlavní část vývoje T lymfocytů probíhá v thymu. Vznikají dvě hlavní, fenotypicky odlišné populace: prekurzory cytotoxických T (Tc) a pomocných T buněk (Th). B a T buňky se po setkání s antigenem diferencují na efektorové buňky. Část z nich se mění na buňky paměťové, zásadní pro sekundární, anamnestickou odpověď (rychlejší a efektivnější) [9].
Pro výstup buněk z lymfatických uzlin jsou důležité chemokiny a sfingosin. Zablokování receptoru pro sfingosin fosfát (S1PR) zabrání výstupu paměťových T lymfocytů ze sekundárních lymfatických tkání, a tak i jejich vstupu do CNS. Na tomto principu funguje fingolimod (blokuje S1PR), který redukuje počet periferních lymfocytů (vč. potenciálně autoreaktivních T a B buněk) [10,11].
Součástí migrace do místa zánětu (CNS) je přechod přes hematoencefalickou bariéru (HEB). Fyziologicky je prostupná minimálně, ale zvyšuje se za přítomnosti prozánětlivých cytokinů. Při přechodu přes HEB dochází k adhezi buněk na endotel (především pomocí molekul LFA1/ ICAM1 a VLA4/ VCAM1). Natalizumab (monoklonální protilátka reagující s VLA4) tento proces blokuje. Následuje překonání bazální membrány a vrstvy výběžků astrocytů pomocí proteolytických enzymů, které tyto migrující buňky produkují (především matrixové metaloproteinázy) [12].
Překvapivým poznatkem je, že podle Cao et al mají pacienti s RS podobný počet cirkulujících myelin-specifických T buněk. Jejich význam v patogenezi onemocnění mají zřejmě funkční rozdíly těchto lymfocytů, které produkují více prozánětlivých (IL-17) než protizánětlivých (IL-10) cytokinů (na rozdíl od zdravých kontrol, kde je tento poměr opačný). Zajímavé je, že ani u zdravých lidí nejsou tyto buňky naivní, naopak byly již aktivovány a tato aktivace u nich vedla k sekreci inhibičních působků (IL-10) [13].
B lymfocyty
B lymfocyty tvoří malou část zánětlivých buněk v lézích pacientů s RS. Proto byl jejich význam dlouho podceňován. Prvním vodítkem byl již objev oligoklonálních imunoglobulinů v likvoru, důkaz intratékální syntézy specifických protilátek. Jejich antigeny, zatím neznámé, by mohly najít využití v diagnostice a monitoraci aktivity onemocnění [14].
Existuje několik mechanizmů účinku B lymfocytů v patogenezi RS. Protilátky působí protilátkovou cytotoxicitou a aktivací komplementu. Dalšími mechanizmy jsou transport a prezentace (auto)antigenu T lymfocytům, poskytnutí kostimulačních signálů přímou B– T interakcí a produkce cytokinů (IL-6).B lymfocyty, které působí protizánětlivě (např. produkcí IL-10 a IL-35), se nazývají regulační, Breg, neboli B10. Usměrňují funkci makrofágů a dendritických buněk, snižují proliferaci CD4+ buněk a zvyšují expresi Treg [15,16].
Při vývoji B lymfocytů je žádoucí velká diverzita B buněčných receptorů. Rizikem je ale vznik autoagresivních klonů. Důležitou roli hraje eliminace těchto klonů na dvou úrovních: centrální tolerance (v kostní dřeni) a periferní tolerance (po jejím opuštění). Některé studie prokázaly ztrátu této B buněčné autotolerance u pacientů s RS [17].
Aktivované B lymfocyty mají znak CD20. Po selektivní depleci B lymfocytů pomocí monoklonální protilátky proti CD20 (rituximab) došlo k redukci relapsů u pacientů s RS. Stejně tak při plazmaferéze nebo imunoadsorpci [18].
Potenciál terapie zaměřené na B lymfocyty spočívá v několika cestách:
- deplece B lymfocytů pomocí monoklonální protilátky proti CD20 (rixutimab, ofatumumab, ocrelizumab), nebo proti CD52 (alemtuzumab);
- deplece plazmatických buněk (alemtuzumab, anti-IL6/ IL-6R, atacicept, bortezomib);
- neutralizace BAFF (B-cell Activating Factor) a APRIL (A Proliferation-Inducing Ligand), cytokinů důležitých pro maturaci a přežití B lymfocytů vč. autoreaktivních klonů (atacicept, tabalumab, belimumab);
- omezení transportu antigenu vychytávaného z cirkulace (fingolimod) [15,19].
Některé z těchto možností jsou zobrazeny na obr. 1 [20].

T lymfocyty
Vývoj a struktura T lymfocytů
T lymfocyty se vyvíjí převážně v thymu z prothymocytů, které tam vycestovaly z kostní dřeně během prenatálního vývoje. Dochází k jejich selekci a buňky, které přežijí (cca 5 %), vstupují do krevního oběhu a dále do periferních lymfatických orgánů [21].
V kortexu thymu probíhá tzv. pozitivní selekce, přežívají pouze buňky schopné vázat MHC molekuly vlastní danému jedinci (asi 10– 30 %). Dále navazuje v části kortikomedulární tzv. negativní selekce, kdy jsou eliminovány potenciálně autoreaktivní buňky, které váží MHC molekuly s vlastními peptidy vysokou afinitou [22].
Po migraci do periferních lymfatických orgánů se T lymfocyty setkávají s antigeny, jsou aktivovány a jejich úkolem je eliminace potenciálních patologických agens (bakterií, virů, nádorových buněk).
Na povrchu T lymfocytů nacházíme znak CD3 a další podle jednotlivých subtypů, CD8 cytotoxické, CD4 pomocné, CD25 regulační. V RS plakách se CD4+ buňky vyskytují především v perivaskulárních prostorech a meningách, zatímco CD8+ v parenchymu [23]. Některé z možných cest vývoje naivního T lymfocytu jsou ilustrovány na obr. 2 [24].
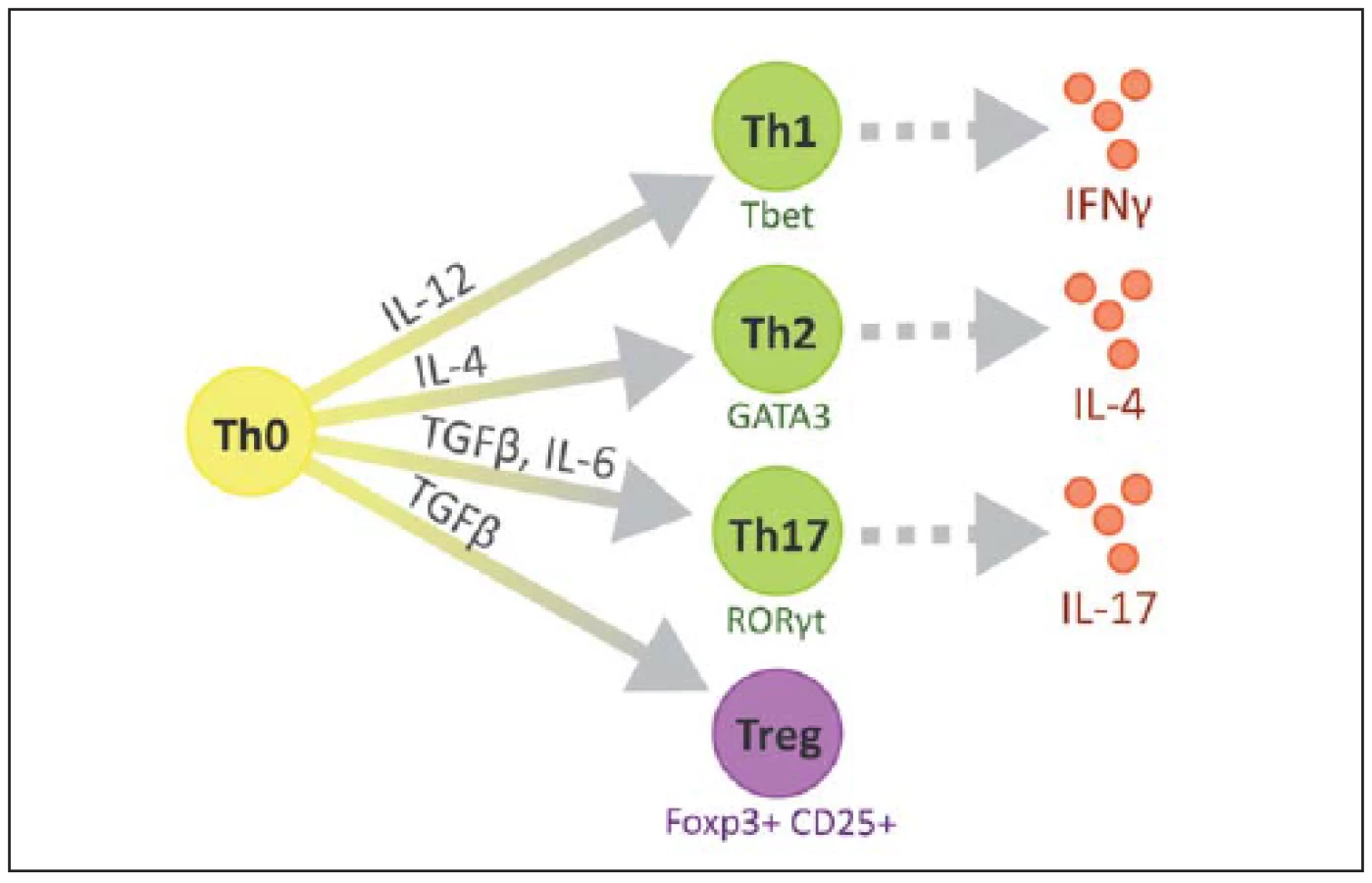
Jednotlivé typy T lymfocytů
Cytotoxické T lymfocyty
Tc ochraňují CNS před infekčními agens a poškozenými vlastními buňkami (vč. neoplasticky změněných). Na povrchu nesou molekuly CD8, pomocí kterých rozeznávají komplexy MHC glykoproteinů s antigeny na povrchu APC (Antigen-Presenting Cell). Tc navozují apoptotickou smrt nakažené buňky (např. pomocí perforinů a granzymů, přes povrchový receptor Fas, CD95, nebo produkcí TNF-β) [25].
Tc se významně podílí i na patogenezi RS. Během probíhajícího zánětu a těsně po akutní demyelinizaci jsou axony vulnerabilní, dochází ke ztrátě schopnosti vedení elektrického impulzu, následuje přechodná exprese molekul MHC I, a vlákno se tak stává terčem útoku Tc a makrofágů [3].
Mikromanipulace a jednobuněčné PCR studie prokázaly, že Tc jsou dominující složkou T buněčného infiltrátu aktivních lézí CNS, ale i převažujícím lymfocytárním typem ve všech stadiích RS [25,26].
Některé typy Tc pravděpodobně hrají v demyelinizačních lézích roli regulační, tedy potlačující zánět [27].
Lékem inhibujícím aktivitu Tc je takrolimus (FK 506), neselektivní imunosupresivum [28].
Pomocné T lymfocyty
Th na svém povrchu exprimují CD4 glykoprotein. Tento subtyp je aktivován vazbou molekuly MHC II. Naivní CD4+ lymfocyty se mohou diferencovat v pomocné (Th1, Th2, Th9, Th17) nebo regulační lymfocyty (Treg). Th1 a Th17 hrají roli v imunitní odpovědi proti intra- a extracelulárním patogenům. Jejich přílišná aktivita může způsobit autoimunitní nebo zánětlivé onemocnění. Treg navozují imunitní toleranci a potlačující zvýšenou aktivitu Th [29].
Jednotlivé typy Th lze rozlišit podle účinku a převažujících cytokinů, které produkují, na Th1, jež mají v jádře transkripční faktor T-bet a produkují zejména IL-1 a IFNγ. Th2 mající v jádře transkripční faktor GATA-3 a produkující hlavně IL-4, IL-5, IL-6 a IL-10. Tzv. Th0 klony produkují směs cytokinů typu 1 a 2, Th3 produkují TGFβ, Tr1 produkují IL-10 a Th17 produkují IL-17, který má výrazně prozánětlivé vlastnosti. Tyto buňky se diferencují z Th0 pod vlivem IL-23 (produkovány buňkami nespecifické imunity) a produkují i IL-21, který aktivuje NK buňky [29,30].
V patogenezi RS hrají roli i tzv. mikroRNA, což jsou malé nekódující molekuly RNA. Dle autorů Zhang et al je několik typů mikroRNA podílejících se na regulaci vývoje imunitních buněk a jejich odpovědí. Z nich má největší význam MiR-155, která ovlivňuje funkci patologických buněk, jako např. T a B buněk a dentritických buněk. Zvýšením jeho exprese dochází ke zvýšení počtu Th1 a Th17 lymfocytů, a tím zhoršení průběhu EAE a naopak. Jde tedy o potenciální diagnostický a prognostický marker a zároveň možný terapeutický cíl [31].
Th1
Th1 působí prostřednictvím aktivace Tc, NK buněk a aktivace makrofágů prostřednictvím produkce IFNγ a IL-2, IL-12, IL-18. IFNγ aktivuje mozkový endotel a zvyšuje expresi specifických adhezních molekul na povrchu endotelových buněk HEB [30,32].
Makrofágy a dendritické buňky produkují IL-12, který je diferenciačním faktorem pro Th1 (přes transkripční faktor STAT4, signal transducer and activator of transcription). Účinek Il-12 posilují IL-18 a TNFα. Zároveň působí negativně na diferenciaci směrem ke klonům Th2 (obdobně působí i cytokiny pro diferenciaci Th2). Význam má i IL-2, který jako autokrinní růstový faktor podporuje proliferaci Th1 [4,33].
Za patologických podmínek zesílená aktivita subsetu Th1 vede ke vzniku imununopatologické reakce IV. typu, charakterizované akumulací Th1 a aktivovaných makrofágů vytvářejících tzv. granulom. Tento typ reakce je prokazován v lézích CNS nemocných s RS. Dříve byly Th1 považovány za hlavní patogenetický činitel při rozvoji RS, proto je řada léků zaměřena na potlačení jejich aktivity [12,25].
Th2
Základními produkty Th2, mířících proti extracelulárním patogenům, jsou především IL-4, IL-5, IL-6, IL-9 a IL-10. K diferenciaci je nutné setkání prekurzorové buňky s antigenem (komplexem MHC gp II a antigenního peptidu) na povrchu APC za přítomnosti IL-4 (produkován bazofily a mastocyty, působí přes signální protein STAT 6) [34].
Th2 jsou protizánětlivé. Mechanizmem je stimulace B lymfocytů (již antigenně stimulovaných) k produkci protilátek [4,35]. Tohoto účinku využívá glatiramer acetát (GA), který aktivuje vznik specifických GA reaktivních Th2 buněk. Ty putují přes HEB do CNS, kde potlačují prozánětlivou aktivitu Th1. Tyto specifické Th2 také secernují neurotrofiny důležité pro přežití axonů a ochranu axonů. GA má pozitivní vliv na oligodendrocyty, jež mohou přispívat k reparaci myelinu [36,37].
Th17
Th17 jsou novým typem lymfocytů, objeveným v roce 2005. Jsou charakterizovány produkcí cytokinu IL-17, ale i TNFα a IL-22. Pro jejich diferenciaci jsou nezbytné TGFβ a IL-6. TGFβ je důležitý pro udržení imunologické tolerance. V nízkých koncentracích indukuje expresi Th17, ve vysokých naopak expresi Treg. Tyto cytokiny působí přes transkripční faktory STAT3 a RORγ a RORα a další (např. NF-kB, IkBf, BATF). Na vývoji Th17 se podílí i IL-23, důležitý pro expanzi a stabilitu fenotypu Th17 a IL-21 pro amplifikaci odpovědí Th17 [38– 40]. Cytokiny IFNγ a IL-4 inhibují jejich diferenciaci. Th17 mohou pravděpodobně vznikat i z efektorových paměťových Th. Podle některých studií je možné rozdělit Th17 podle typu diferenciace na: 1. TGF-nezávislé vzniklé Th17, „nepatogenní“ a na 2. TGF-závislé Th17, „patogenní“. IL-6 je pleiotropní cytokin secernovaný buňkami vrozeného imunitního systému, B a T buňkami, nádorovými buňkami, fibroblasty, endotelovými buňkami a keratinocyty. Má roli v regulaci rovnováhy Th17 a Treg. Indukuje diferenciaci Th17, zatímco inhibuje vznik Treg [41].
Th17 jsou vysoce prozánětlivé a prostřednictvím produkce IL-17A a IL-17F vedou k náboru a migraci neutrofilů a makrofágů do místa zánětu a k expanzi myeloidních, dentritických i T buněk. Dále indukují produkci prozánětlivých cytokinů, chemokinů, antimikrobiálních peptidů a matrix metaloproteináz [41]. Pozitivní zpětnou vazbou také zvyšují zapojení dalších Th17 do zánětlivého procesu. Napomáhají transmigraci CD4+ lymfocytů přes HEB pomocí narušování mezibuněčných spojů typu tight junctions (IL-17) a působí synergicky s INFγ v indukci exprese ICAM1 na endotelu [22]. Spolu s Th1 jsou hlavními prozánětlivými T buňkami. Th17 se pravděpodobně uplatňují v počátečních fázích onemocnění, naproti tomu Th1 až v pozdějších. Hrají tak významnou roli v obraně proti extracelulárním a mykotickým patogenům. Jsou také součástí slizniční bariéry. Při jejich depleci dochází k chronickým slizničním zánětům. Jako jejich další produkty byly identifikovány IL-21 a IL-22. IL-21 působí autokrinně, jeho exprese je závislá na přítomnosti IL-6 a působí synergicky s TGFβ, podporuje diferenciaci Th17. Naopak IL-22 se váže na receptory cílových buněk a indukuje expresi antimikrobiálních peptidů (β-defensin-2, β-defensin-3) [28]. Babaloo et al svou studií potvrdili pozitivní korelaci mezi sérovou hladinou IL-17 a počtem relapsů. Naopak nebyl nalezen vztah mezi tímto cytokinem a mírou disability (EDSS) ani progresí onemocnění [42].
Byly popsány i buňky produkující zároveň IFNγ i IL-17. Zvažuje se, zda jde o nový typ T buněk, nebo přechodovou formu mezi nestabilními Th17 a Th1. O diferenciaci v Th17 nebo Th1 rozhoduje přítomnost TGFβ [29].
Všechny prozánětlivé aktivity Th17 jsou regulovány jiným imunoregulačním subsetem lymfocytů, Treg lymfocyty [11].
Klony Th17 specificky namířené proti autoantigenům jsou spojovány s autoimunitními onemocněními. Společnou charakteristikou těchto onemocnění je chronický nebo rekurentní průběh. Souvislost s RS je zjevná, při ní dentritické buňky produkují více IL-23, ale normální množství IL-12 ve srovnání se zdravými jedinci. Microarray analýza plak získaných autopsií prokázala zvýšení IL-17 mRNA ve srovnání se vzorky pacientů bez patologie CNS. Ukázalo se, že Th17 lymfocyty efektivněji přestupují HEB než Th1. IL-17 deficientní zvířecí modely vyvinou EAE opožděně a onemocnění probíhá méně závažně. Při pokusech s protilátkou namířenou proti IL-12 a IL-23 provedených na hlodavcích a primátech došlo k zabránění vzniku EAE. Použití této neutralizační protilátky (ustekinumab) nebylo u pacientů s RR (relabující-remitentní) RS úspěšné [38– 40]. Byl proveden rozbor krve a mozkomíšního moku pacientů s RR RS, CIS (klinicky izolovaný syndrom; obojí během relapsu a remise), zdravých kontrol a pacientů s jiným zánětlivým onemocněním CNS. V periferní krvi byl počet Th17 nízký, bez signifikantního rozdílu mezi skupinami. Naopak v mozkomíšním moku byl počet Th17 vyšší u pacientů s jiným zánětlivým onemocněním CNS a RRRS při relapsu. Počet Th17 při relapsu byl významně vyšší než v remisi. Počet Th17 byl v likvoru asi dvojnásobný oproti periferní krvi v období relapsu i remise. Vztah mezi počtem Th17 a disabilitou nebo trváním onemocnění nebyl ani v této studii zaznamenán. V absolutním počtu v krvi i v likvoru převažovaly Th1 (50 %) nad Th17 (2–6 %), avšak pouze zvýšení hladiny Th17 odráželo probíhající relaps [23].
Li et al zkoumali vliv vysokých dávek intravenózních glukokortikoidů podaných během relapsu (u pacientů s RS a neuromyelitis optica) na počet Th17 a hladiny cytokinů. Po přeléčení 3 g metylprednizolonu došlo k signifikantnímu snížení počtu Th17 i snížení hladiny IL-17 a IL-23. Přitom Th17 a sérová hladina IL-17 korelovala s EDSS skóre a hladina IL-23 byla nepřímo úměrná trvání onemocnění [43].
Dalším proteinem nezbytným pro diferenciaci a funkčnost Th17 a také pro produkci IL-17 je protein p38 MAPK (Mitogen-Activated Protein Kinase). U pacientů s RR RS vykazují tyto buňky pozměněnou odpovídavost v průběhu p38 signální dráhy, která vede ke zvýšené fosforylaci proteinu p38. Ten je aktivován stresem, zánětlivými signály (např. TNFα aj.) a alternativní cestou pomocí TCR (T-Cell Receptor). Farmakologická inhibice p38 u myší vedla ke zmírnění průběhu EAE. Novou perspektivou léčby je užití inhibitorů p38, které mají potenciál ovlivnit jak hladinu Th17, tak i produkci IL-17 z již diferencovaných Th17. Nevýhodou je jejich potenciální neuro- a hepatotoxicita [44].
Regulační T lymfocyty
Pomáhají udržovat imunologickou toleranci. Potlačují imunitní reakce, omezují aktivaci, proliferaci, přežívání a prozánětlivé účinky různých imunitních buněk vč. Th. Tvoří 5– 10 % CD4+ T lymfocytů. Při jejich depleci u jinak normálních myší dochází ke vzniku autoimunitních onemocnění [33,38].
Treg utlumují T buněčnou imunitní reakci a potlačují autoreaktivní T lymfocyty, které prošly negativní selekcí v thymu. Jsou charakteristické expresí znaku CD25, který ale není specifický. Mohou být identifikovány pomocí jaderného transkripčního faktoru Foxp3 (Forkhead box protein 3). Mutace genu pro Foxp3 způsobuje IPEX (Immune dysregulation, Polyendocrinopathy, Enteropathy, X-linked) fatální lymfoproliferativní poruchu vedoucí k multiorgánovému selhání [29].
Podle jejich vzniku, fenotypu a způsobu účinku můžeme rozlišit několik subtypů Treg. Foxp3 je hlavním transkripčním faktorem většiny z nich. Rozeznáváme 1. nTreg (natural), Foxp3+, vyvíjející se v thymu, 2. iTreg (inducible), Foxp3+, vyvíjí se v periferních tkáních, 3. Tr1, Foxp3–, secernující IL-10, 4. Th3 produkující TGF-β (obr. 3) [45]. Oba hlavní typy (nTreg a iTreg) exprimují podobné molekuly, např. CD25, CD122, CTLA-4, GITR, CCR4, CD62L, PD1 a Foxp3 a CD45R0 [29].
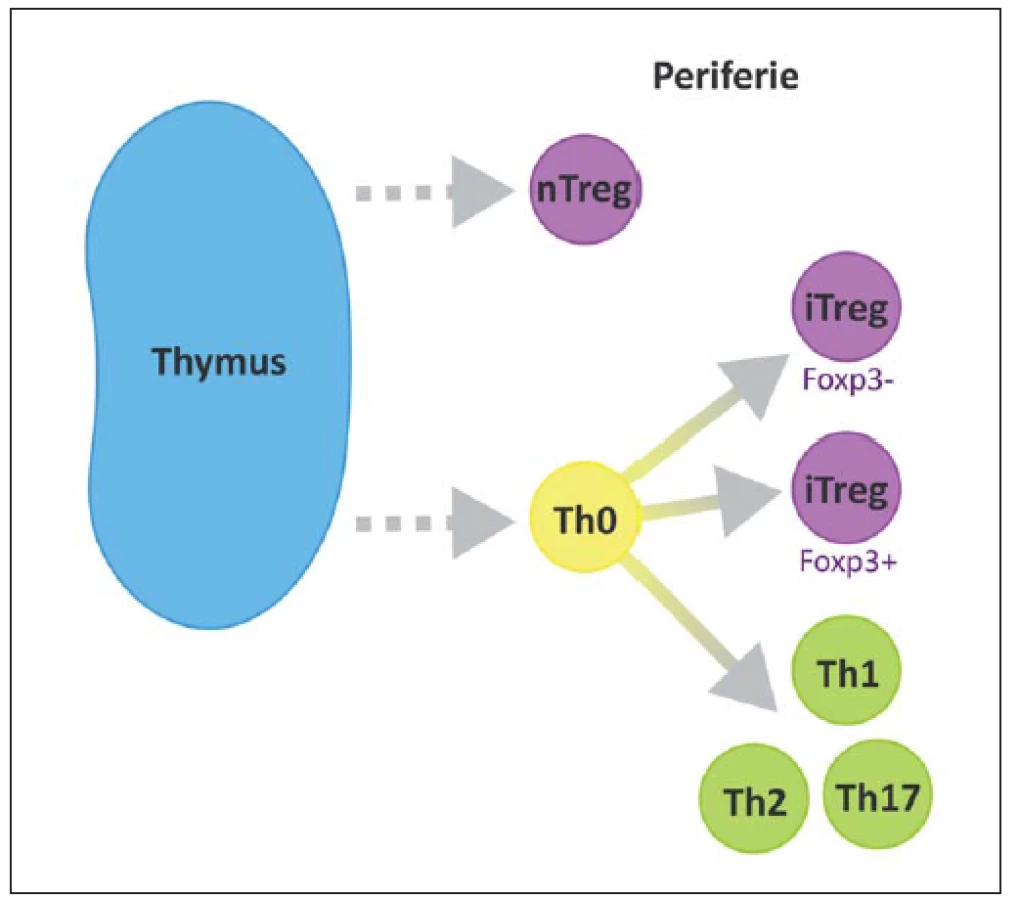
Od roku 1994, kdy Tonegawa et al popsali možný protektivní efekt Treg, proběhlo několik studií na animálních modelech, všechny s podobným výsledkem prokazujícím pozitivní účinky Treg při rozvoji EAE [46].
nTreg se vyvíjejí v thymu pomocí rozpoznání vlastních antigenů, při jejich vývoji je nutná interakce MHC molekuly a TCR. Zároveň je nutná kostimulace pomocí CD28. IL-2 a TGFβ nejsou nezbytné [47]. nTreg tvoří 5– 10 % všech CD4+ thymocytů a 10 % CD4+ periferních buněk [16].
Naopak vývoj iTreg je závislý na přítomnosti TGFβ, funkčnosti jeho receptoru a na IL-2. Není potřeba CD28, mohou být Foxp3– [38]. iTreg se vyvíjí především na sliznicích [16].
Dlouhou dobu bylo velmi složité od sebe nTreg a iTreg odlišit. Dnes se tak děje pomocí produkovaného neuropilinu (iTreg jej produkují v daleko menším množství). I funkce obou subtypů je odlišná. nTreg snižují aktivitu autoreaktivních T buněk (těch, které unikly destrukci v thymu), naproti tomu iTreg zabraňují aktivitě efektorových T buněk (indukované během imunitní odpovědi na antigen). Jejich význam proto není zaměnitelný, jsou navzájem komplementární [16].
Podle mnohých studií se ukazuje, že počty aktivovaných lymfocytů a Treg jsou snížené u autoimunitních nemocí. Závažnost a aktivita onemocnění úměrně odpovídá stupni jejich deficitu. Jiné studie však toto vyvrací. Pravděpodobně nemusí jít pouze o snížený počet těchto buněk, ale i poruchu jejich funkce. Zde spočívá potenciál zaměření budoucí léčby [48].
Deplece Treg usnadňuje vznik Th17, a tím i náchylnost myší k EAE. Podle Stephense et al zvyšují práh pro vznik autoreaktivních odpovědí a takto i snižují riziko vzniku autoimunitního onemocnění. Dochází k tomu prostřednictvím IL-10, jak bylo ukázáno na animálních modelech (kdy u IL-10 depletních myší nedošlo k potlačení EAE). Většina Treg se v CNS nachází během zotavovací fáze, kdy tvoří až jednu třetinu CD4+ buněk. Jejich deplece zabraňuje zotavení. Liu et al uvádí, že neurony mohou indukovat konverzi myelin-specifických autoagresivních efektorových T buněk v Treg s jejich supresivní aktivitou [29,38].
O významu Treg v patogenezi RS se uvažuje od chvíle, kdy byly tyto buňky se sníženou supresivní aktivitou nalezeny u pacientů s RS. Zajímavé je, že u pokročilejších fází onemocnění sekundárně progresivní formy RS měly Treg normální supresivní funkci. Tento funkční defekt může být korigován terapií IFNβ a copolymerem-I [32].
Závěr
Na vzniku a rozvoji RS se podílí všechny složky imunity. Zásadní roli hrají buňky imunity specifické. Z nich jsou v centru pozornosti především Th1 a Th2 a v posledním desetiletí nově také Th17 a Treg. Procesu se účastní i B lymfocyty, buňky vrozené imunity (makrofágy, dendritické buňky aj.) a své místo má samozřejmě i humorální imunita (cytokiny, chemokiny, protilátky aj.). Velmi pravděpodobný je také budoucí objev dalších subpopulací B a T lymfocytů a jiných buněk imunitního systému.
Poznání všech těchto a mnohých dalších patogenetických procesů účastnících se vzniku a vývoje RS povede k lepším terapeutickým možnostem a cílené terapii s méně nežádoucími účinky.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr. Monika Svobodová
Neurologická klinika
LF MU a FN Brno
Jihlavská 20
625 00 Brno
email: svobodova.monika@fnbrno.cz
Přijato k recenzi: 22. 9. 2016
Přijato do tisku: 5. 1. 2017
Zdroje
1. Havrdová E. Neuroimunologie. 1. vyd. Praha: Maxdorf 2001.
2. Constantinescu CS, Farooqi N, O’Brien K, et al. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br J Pharmacol 2011;164(4):1079– 106. doi: 10.1111/ j.1476-5381.2011. 01302.x.
3. Kuhlmann T, Lassmann H, Brück W. Diagnosis of inflammatory demyelination in biopsy specimens: a practical approach. Acta Neuropathol 2008;115(3)275– 87. doi: 10.1007/ s00401-007-0320-8.
4. Lauerová L, Kocák I. Regulace protinádorové imunity pomocnými CD4+ TH1/ TH2 lymfocyty. Klin Onkol 2001;14(5):154– 6.
5. Noble A, Thomas MJ, Kemeny DM. Early Th1/ Th2 cell polarization in the absence of IL-4 and IL-12: T cell receptor signaling regulates the response to cytokines in CD4 and CD8 T cells. Eur J Immunol 2001;31(7):2227– 35.
6. Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol 2004;22:745– 63. doi: 10.1146/ annurev.immunol.22.012703.104702.
7. Sakaguchi S, Yamaguchi T, Nomura T, et al. Regulatory T cells and immune tolerance. Cell 2008;133(5):775– 87.doi: 10.1016/ j.cell.2008.05.009.
8. Acosta-Rodriguez EV, Rivino L, Geginat J, et al. Surface phenotype and antigenic specificity of human interleukin 17 producing T helper memory cells. Nat Immunol 2007;8(6):639– 46. doi: 10.1038/ ni1467.
9. Horejší V, Bartůňková J. Základy imunologie. 4. vyd. Praha: Triton 2009.
10. Chiba K, Adachi K. Discovery of fingolimod, the sphingosine 1 phosphate receptor modulator and its application for the therapy of multiple sclerosis. Future Med Chem 2012;4(6):771– 81. doi: 10.4155/ fmc.12.25.
11. Sato DK, Nakashima I, Bar-Or A, et al. Changes in Th17 and regulatory T cells after fingolimod initiation to treat multiple sclerosis. J Neuroimmunol 2014;268(1– 2):95– 8. doi: 10.1016/ j.jneuroim.2014.01.008.
12. Yusuf-Makagiansar H, Anderson ME, Yakovleva TV, et al. Inhibition of LFA-1/ ICAM-1 and VLA-4/ VCAM-1 as a therapeutic approach to inflammation and autoimmune diseases. Med Res Rev 2002;22(2):146– 67.
13. Mehling M, Johnson TA, Antel J, et al. Clinical immunology of the sphingosine 1 phosphate receptor modulator fingolimod (FTY720) in multiple sclerosis. Neurology 2011;76(Suppl 3):S20– 7. doi: 10.1212/ WNL.0b013e31820db341.
14. Cao Y, Goods BA, Raddassi K, et al. Functional inflammatory profiles distinguish myelin-reactive T cellsfrom patients with multiple sclerosis. Sci Transl Med2015;7(287):287ra74. doi: 10.1126/ scitranslmed.aaa8038.
15. Villar LM, Masterman T, Casanova B, et al. CSF oligoclonal band patterns reveal disease heterogeneity in multiple sclerosis. J Neuroimmunol 2009;211(1– 2):101– 4. doi: 10.1016/ j.jneuroim.2009.03.003.
16. Ireland S, Monson N. Potential impact of B cells on T cell function in multiple sclerosis. Mult Scler Int 2011;2011:423971. doi: 10.1155/ 2011/ 423971.
17. Buc M. Role of regulatory T cells in pathogenesis and biological therapy of multiple sclerosis. Mediators Inflamm 2013;2013:963748. doi: 10.1155/ 2013/ 963748.
18. Tobón GJ, Izquierdo JH, Cañas CA. B lymphocytes: development, tolerance, and their role in autoimmunity-focus on systemic lupus erythematosus. Autoimmune Dis 2013;2013:827254. doi: 10.1155/ 2013/ 827254.
19. Cross AH, Klein RS, Piccio L. Rituximab combination therapy in relapsing multiple sclerosis. Ther Adv Neurol Disord 2012;5(6):311– 9. doi: 10.1177/ 1756285612461165.
20. Krumbholz M, Meinl E. B cells in MS and NMO: pathogenesis and therapy. Semin Immunopathol 2014;36(3):339– 50. doi: 10.1007/ s00281-014-0424-x.
21. Scollay R, Smith J, Stauffer V. Dynamics of early T cells:prothymocyte migration and proliferation in the adult mouse thymus. Immunol Rev 1986;91:129– 57.
22. Starr TK, Jameson SC, Hogquist KA. Positive and negative selection of T cells. Annu Rev Immunol 2003; 21:139– 76. doi: 10.1146/ annurev.immunol.21.120601.141107.
23. Brucklacher-Waldert V, Stuerner K, Kolster M, et al. Phenotypical and functional characterization of T helper 17 cells in multiple sclerosis. Brain 2009;132(12):3329– 41. doi: 10.1093/ brain/ awp289.
24. Neumann H, Medana IM, Bauer J, et al. Cytotoxic T lymphocytes in autoimmune and degenerative CNS diseases. Trends Neurosci 2002;25(6):313– 9.
25. Lassmann H, Ransohoff RM. The CD4-Th1 model for multiple sclerosis: a critical [correction of crucial] re-appraisal. Trends Immunol 2004;25(3):132– 7. doi: 10.1016/ j.it.2004.01.007.
26. Denic A, Wootla B, Rodriguez M. CD8+ T Cellsin Multiple Sclerosis. Expert Opin Ther Targets 2013;17(9):1053– 66. doi: 10.1517/ 14728222.2013.815726.
27. Lake DB, Poole TR. Tacrolimus. Br J Ophthalmol 2003;87(1):121– 2.
28. Zheng SG. Regulatory T cells vs Th17: differentiation of Th17 versus Treg, are the mutually exclusive? Am J Clin Exp Immunol 2013;2(1):94– 106.
29. Lovett-Racke AE, Yang Y, Racke MK. Th1 versus Th17: are T cell cytokines relevant in multiple sclerosis? Biochim Biophys Acta 2011;1812(2):246– 51. doi: 10.1016/ j.bbadis.2010.05.012.
30. Zhang J, Cheng Y, Cui W, et al. MicroRNA-155 modulates Th1 and Th17 cell differentiation and is associated with multiple sclerosis and experimental autoimmune encephalomyelitis. J Neuroimmunol 2014;266(1– 2):56– 63. doi: 10.1016/ j.jneuroim.2013.09.019.
31. Kebir H, Ifergan I, Alvarez JI, et al. Preferential recruitment of interferon-gamma-expressing TH17 cells in multiple sclerosis. Ann Neurol 2009;66(3):390– 402. doi: 10.1002/ ana.21748.
32. Rostami A, Ciric B. Role of Th17 cells in the pathogenesis of CNS inflammatory demyelination. J Neurol Sci 2013;333(1– 2):76– 87. doi: 10.1016/ j.jns.2013.03.002.
33. Krejsek J, Holmannová D. Imunopatogeneze roztroušené sklerózy mozkomíšní. Postgrad Med 2012;14:933– 8.
34. Maier E, Duschl A, Horejs-Hoeck J. STAT6-dependent and -independent mechanisms in Th2 polarization. Eur J Immunol 2012;42(11):2827– 33. doi: 10.1002/ eji.201242433.
35. Krejsek J, Kopecký O. Klinická imunologie. 1. vyd. Hradec Králové: NUCLEUS HK 2004.
36. Liblau R. Glatiramer acetate for the treatment of multiple sclerosis: evidence for a dual anti-inflammatory and neuroprotective role. J Neurol Sci 2009;287(Suppl 1):S17– 23. doi: 10.1016/ S0022-510X(09)71296-1.
37. Schrempf W, Ziemssen T. Glatiramer acetate: mechanisms of action in multiple sclerosis. Autoimmun Rev 2007;6(7):469– 75. doi: 10.1016/ j.autrev.2007.02.003.
38. Bettelli E, Korn T, Kuchroo VK. Th17: the third member of the effector T cell trilogy. Curr Opin Immunol 2007;19(6):652– 7. doi: 10.1016/ j.coi.2007.07.020.
39. Miossec P. IL-17 and Th17 cells in human inflammatory diseases. Microbes Infect 2009;11(5):625– 30. doi: 10.1016/ j.micinf.2009.04.003.
40. Lee YK, Mukasa R, Hatton RD, et al. Developmental plasticity of Th17 and Treg cells. Curr OpinImmunol 2009;21(3):274– 80. doi: 10.1016/ j.coi.2009.05.021.
41. Zambrano-Zaragoza JF, Romo-Martínez EJ, Durán-Avelar MJ, et al. Th17 cells in autoimmune and infectious diseases. Int J Inflamm 2014;2014:651503. doi: 10.1155/ 2014/ 651503.
42. Babaloo Z, Aliparasti MR, Babaiea F, et al. The role of Th17 cells in patients with relapsing-remitting multiple sclerosis: interleukin-17A and interleukin-17F serum levels. Immunol Lett 2015;164(2):76– 80. doi: 10.1016/ j.imlet.2015.01.001.
43. Li Y, Wang H, Long Y, et al. Increased memory Th17 cells in patients with neuromyelitis optica and multiple sclerosis. J Neuroimmunol 2011;234(1– 2):155– 60. doi: 10.1016/ j.jneuroim.2011.03.009.
44. Di Mitri D, Sambucci M, Loiarro M, et al. The p38 mitogen-activated protein kinase cascade modulates T helper type 17 differentiation and functionality in multiple sclerosis. Immunology 2015;146(2):251– 63. doi: 10.1111/ imm.12497.
45. Jamshidian A, Shaygannejad V, Pourazar A, et al. Biased Treg/ Th17 balance away from regulatory toward inflammatory phenotype in relapsed multiple sclerosis and its correlation with severity of symptoms. J Neuroimmunol 2013;262(1– 2):106– 12. doi: 10.1016/ j.jneuroim.2013.06.007.
46. Poojary KV, Kong YM, Farrar MA. Control of Th2-mediated inflammation by regulatory T cells. Am J Pathol 2010;177(2):525– 31. doi: 10.2353/ ajpath.2010.090936.
47. Dejaco C, Duftner C, Grubeck-Loebenstein B, et al. Imbalance of regulatory T cells in human autoimmune diseases. Immunology 2006;117(3):289– 300. doi: 10.1111/ j.1365-2567.2005.02317.x.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2017 Číslo 2
Nejčtenější v tomto čísle
- Loketní nerv
- Incidence cévní mozkové příhody v Evropě – systematická review
- Anti-NMDAR encefalitida v dětském věku – kazuistika
- Disekce karotid neantikoagulujeme