
Konformačně specifické protilátky a diagnostika prionových chorob
Conformation Specific Antibodies and Diagnosis of Prion Diseases
Transmissible spongiform encephalopathies, or prion diseases, are fatal neurodegenerative disorders with long incubation period and short clinical phase. The diagnosis is usually confirmed from post mortem detection of prions in the brain tissue. Most diagnostic methods are based on treatment with Proteinase K that cleaves out physiological proteins and makes the resistant form of pathological prion protein detectable with anti-prion antibodies. Over the last five years, there is a growing evidence of prionopathies caused by protease-sensitive prions escaping detection with the standard diagnostic methods. Conformation-specific monoclonal antibodies to the pathological prion proteins could overcome the problem simply by detecting the pathological conformation of both prion isoforms without the need for proteolysis. Our review summarizes available information on conformation-specific prion antibodies and discusses their use in the diagnosis of prion diseases.
Key words:
transmissible spongiform encephalopathy – prion protein – conformation-specific antibodies – Creutzfeldt-Jakob disease
Autoři:
E. Dvořáková; K. Holada
Působiště autorů:
Ústav imunologie a mikrobiologie, 1. LF UK a VFN v Praze
Vyšlo v časopise:
Cesk Slov Neurol N 2012; 75/108(3): 283-290
Kategorie:
Přehledný referát
Souhrn
Transmisivní spongiformní encefalopatie neboli prionová onemocnění jsou smrtelné neurodegenerativní choroby s dlouhou inkubační dobou a rychlým průběhem. K definitivní diagnóze dochází obvykle až post mortem detekcí prionů v mozkové tkáni. Klíčovým krokem diagnostiky prionových chorob bývá použití proteinázy K, která rozštěpí fyziologické proteiny a umožní následnou imunochemickou detekci proteáza-rezistentního prionového proteinu. V posledních pěti letech se celosvětově objevuje stále více případů prionopatií zapříčiněných proteáza-senzitivními priony, které jsou běžnými metodami vyhodnocovány falešně negativně. Řešením problému by mohla být příprava diagnostického testu založeného na použití monoklonálních protilátek konformačně specifických pro patologický prionový protein, které by umožnily potvrzení diagnózy bez použití proteolýzy. Náš text shrnuje dostupné informace o připravených konformačně specifických protilátkách a diskutuje o jejich použití v diagnostice prionových chorob.
Klíčová slova:
transmisivní spongiformní encefalopatie – prionový protein – konformačně specifické protilátky – Creutzfeldtova-Jakobova nemoc
Úvod
Prionové choroby neboli transmisivní spongiformní encefalopatie (TSE) jsou neurodegenerativní onemocnění spojené s úbytkem neuronů, spongiformními změnami, gliózou a ukládáním patologické formy prionového proteinu v mozku [1]. Onemocnění se projevuje například jako klusavka („scrapie“) ovcí a koz, bovinní spongiformní encefalopatie (BSE) skotu nebo chronické chřadnutí (CWD) jelenovitých. První lidská prionová choroba byla popsána v letech 1920 a 1921 Hansem Creutzfeldtem a Alfonsem Jakobem [2,3]. V 50. letech 20. stol. došlo na Nové Guinei k epidemii kuru, která se rozšířila u příslušníků domorodého kmene Fore během rituálního kanibalizmu [4]. Celosvětově je nejčastější lidskou prionovou chorobou Creutzfeldtova-Jakobova nemoc (CJN) s incidencí onemocnění 1–2 případy na milion obyvatel ročně. Onemocnění většinou vzniká bez známých příčin (85 % případů), může ale být i dědičné (10–15 % případů) nebo získané (2–3 % případů). Vzácněji se vyskytuje dědičný Gerstmann-Sträussler-Scheinkerův syndrom (GSS) a fatální familiární insomnie (FFI) [5]. V současnosti velkou pozornost poutá variantní CJN (vCJN), která vznikla s největší pravděpodobností alimentárním přenosem BSE prionů na člověka. Na rozdíl od klasické CJN postihuje především mladé lidi, vCJN priony se akumulují i v orgánech imunitního systému a choroba je přenosná krevní transfuzí [6,7].
Podle prionové hypotézy je infekčním agens prionových chorob patologicky složený prionový protein (PrPTSE, někdy též značený PrPSc), který se množí přímým kontaktem s buněčným prionovým proteinem (PrPC), jemuž dokáže vnutit svoji patologickou, na struktury β skládaného listu bohatou konformaci [1]. PrPC se vyskytuje na povrchu většiny buněk v těle [1,8] a jeho fyziologická funkce zatím nebyla objasněna. Uvažuje se například o jeho úloze v metabolizmu mědi, regulaci apoptózy, v procesu učení a paměti, signální transdukci, přenosu vzruchu na synaptické membráně, ovlivnění cirkadiálního rytmu, buněčné diferenciaci, antioxidační ochraně, neuroprotekčních procesech a dalších buněčných dějích [9]. Molekula PrPC obsahuje ve své sekundární struktuře vysoký podíl α šroubovice, což ji činí dobře štěpitelnou proteázami. Poločas života molekuly PrPC v buňce se odhaduje na 3–6 hod [10,11]. Oproti tomu molekula PrPTSE je v průběhu konformačních změn obohacena o strukturu β skládaného listu (z pouhých 3 % u PrPC na 34 % u PrPTSE) [12,13], která ji činí částečně odolnou vůči proteolýze. PrPTSE navíc agreguje za tvorby amyloidových fibril a vytváří v mozku depozita, která se podílí na rozvoji onemocnění. Konformační změny prionového proteinu zároveň zvyšují afinitu PrPTSE k PrPC, čímž usnadňují další kontakt obou molekul, a tím i propagaci patologické konformace [14,15].
V této fázi již onemocnění nelze zastavit, po dlouhé inkubační době bez příznaků dojde k propuknutí choroby s rychlým koncem. Smrt nastává za 5–7 měsíců po prvním projevu příznaků u většiny sporadických a dědičných případů a za 9–22 měsíců u vCJN. Byla nalezena řada látek schopných interferovat s propagací prionů a odléčit prionem infikované tkáňové kultury [přehled v 16], avšak snahy o vyléčení nebo alespoň pozastavení průběhu onemocnění u lidí se ukázaly být dosud neúspěšné [17,18]. Nejnadějnější výsledky poskytlo použití doxycyclinu, který vedl k signifikantnímu prodloužení doby přežití CJN pacientů – medián 292 dnů (n = 51) oproti 167 dnům u historických kontrol [19]. Vzhledem k identické primární struktuře obou forem prionového proteinu a jen velmi slabé imunogenicitě patologické konformace PrPTSE se v organizmu protilátky specifické pro prionová onemocnění v relevantních titrech netvoří [význam IS u prionových chorob v 20]. Nedávno provedené studie na větším souboru infikovaných myší (143 a 117 jedinců) ukázaly, že jen zhruba 5 % sér odebraných v terminálním stadiu vykazovalo mírně zvýšené titry protilátek proti prionovému proteinu [21]. U lidí se zvýšené titry protilátek proti PrPTSE zatím detekovat nepodařilo.
Diagnostika prionových chorob
Přítomnost PrPTSE je jediným známým specifickým znakem prionových chorob. K diagnostice nelze z výše uvedených důvodů použít běžné sérologické metody. Detekce je ve většině případů založena na rozdílech odolnosti PrPTSE a PrPC vůči štěpení proteinázou K (obr. 1). Zatímco molekula PrPC se působením proteáz zcela rozštěpí, z molekuly PrPTSE se odštěpí pouze N konec a rezistentní C koncová část (PrPres) je imunochemicky detekována některou z protilátek proti prionovému proteinu [přehled detekčních metod v 22]. Tyto testy vycházejí z předpokladu, že rezistentní forma PrPTSE je původcem onemocnění, a tedy i v organizmu přítomna a posléze detekována. V mozku je ale většina molekul PrPTSE proteáza-senzitiv-ních (senPrPTSE), PrPres tvoří pouhých 10–30 % [23–25]. V některých tkáních pacientů s CJN, jako třeba v krvi, se PrPTSE pravděpodobně vyskytuje jen jako senPrPTSE [26]. Detekce PrPTSE v krvi je navíc komplikována přítomností velkého množství PrPC [27–29].
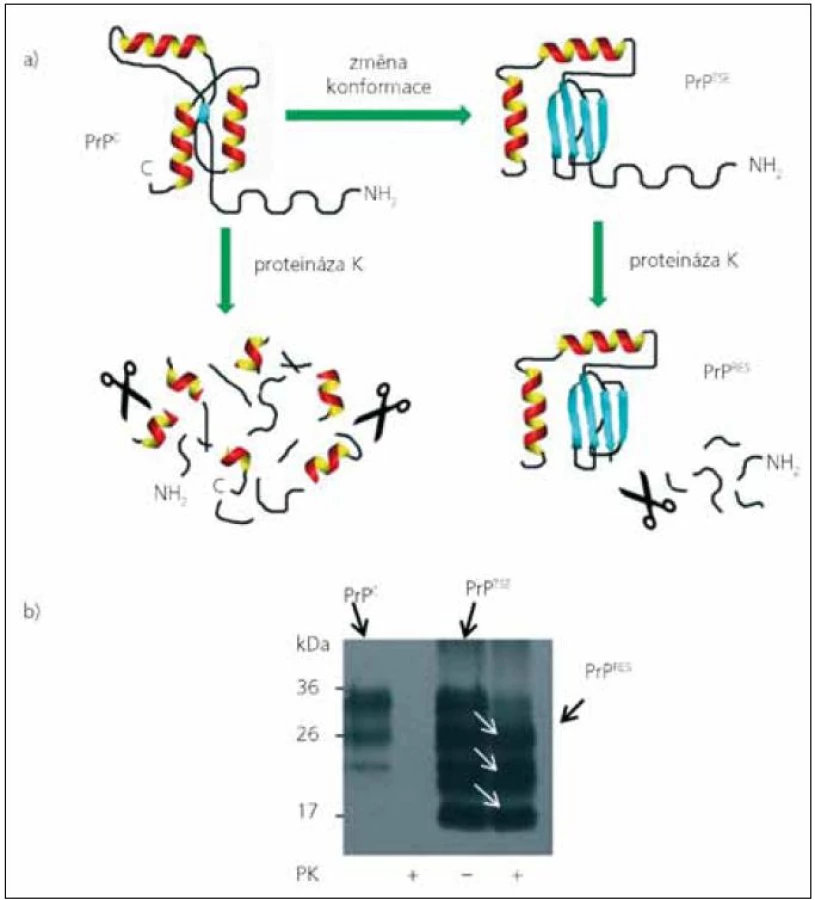
Až donedávna měla prionová komunita za to, že senPrPTSE tvoří mezistupeň při tvorbě PrPres a je součástí průběhu onemocnění [30,31]. SenPrPTSE izolovaný gradientovou centrifugací z infikovaného křeččího mozkového homogenátu byl dokonce schopen konvertovat PrPC na proteáza-rezistentní PrPTSE [32]. V poslední době se ale začínají objevovat případy prionových chorob zvané „proteáza-senzitivní prionopatie“, které mohou, ale nemusejí, mít klinické příznaky podobné klasickým prionovým chorobám [33]. U těchto případů při běžných testech nebyl prokázán PrPres, ale při použití šetrnějších metod byl prokázán senPrPTSE [34–36]. Celosvětově se objevují stále další případy proteáza-senzitivních prionopatií, často po reevaluaci starších případů vyhodnocených jako „blíže nespecifikované demence“ nebo „atypická Alzheimerova choroba“ [37]. Jaká je četnost těchto atypických prionopatií, není v současné době známo.
Problémy s přípravou konformačně specifických protilátek
Nevýhodou diagnostických testů založených na štěpení prionového proteinu proteinázou K je to, že detekci proteáza-senzitivních prionopatií neumožňují. To by umožnila například konformačně specifická protilátka proti PrPTSE, která by zároveň nereagovala s PrPC. Snahy o připravení takové protilátky existují již od 90. let minulého století [38], zdaleka ne vždy se ale setkávají s úspěchem. Problémů s přípravou konformačně specifických protilátek je hned několik. Zaprvé je to slabá imunogenicita PrPTSE, daná shodou primární struktury s fyziologicky se vyskytujícím PrPC. Vzhledem k vysoce mezidruhově konzervativní primární struktuře prionového proteinu tento problém neumožňuje obejít ani xenogenní imunizace. Některým autorům se sice povedlo imunitní reakci podpořit silnými adjuvanty [39,40], problém se nicméně podařilo vyřešit až přípravou geneticky upravených myší, kterým byl odstraněn gen pro prionový protein (Prnp0/0 myši) [41,42] a které po běžné imunizaci vykazovaly dobrou imunitní odpověď. Druhým úskalím při přípravě konformačních protilátek je skutečnost, že dosud není známa prostorová struktura PrPTSE, a nelze tedy s přesností určit epitopy specifické pro patologický prionový protein. Během konformačních změn sice dochází u prionového proteinu k odhalení epitopů, které jsou v molekule PrPC nedostupné [43,44], jejich imunogenicita ovšem nemusí být – a nebývá – vysoká. Po imunizaci Prnp0/0 myší prionovým proteinem tak dojde ke tvorbě celé řady protilátek proti PrP, konformačně specifické se mezi nimi ale běžně nevyskytují [45,46]. Jednotlivé laboratoře se tento problém pokusily řešit různě. Korth et al použili k imunizaci agregovaný rekombinantní prionový protein, který umožnil tvorbu protilátek proti oligomerům PrP, jež se běžně vyskytují v agregátech PrPTSE, ale ne u rozpustného PrPC [38]. Paramithiotis et al a Jones et al imunizovali peptidem molekuly PrP, který vybrali s ohledem na předchozí studie konformačních změn při přechodu z α šroubovice na β helix, k nimž dochází i u PrPTSE [43,47]. Jiné laboratoře zase izolovaly a purifikovaly PrPTSE z mozkového homogenátu infikovaných myší a použily jej jako antigen pro imunizaci a následně i pro selekci protilátek [48,49]. Náš text shrnuje dostupné informace o připravených konformačně specifických protilátkách a diskutuje o jejich použití v diagnostice prionových chorob.
Konformačně specifické protilátky
15B3
První konformačně specifická protilátka byla připravena v roce 1997 imunizací Prnp0/0 myší rekombinantním bovinním prionovým proteinem. Jedná se o monoklonální protilátku 15B3, třídy IgM, která reaguje s bovinním, myším a lidským PrPTSE [38]. 15B3 je schopna z infikovaných mozkových homogenátů imunoprecipitovat PrPTSE různě citlivý ke štěpení proteinázou K. Při bližší charakterizaci epitopu protilátky 15B3 spektrem peptidů byly identifikovány vazebné sekvence 142–148, 162–170 a 214–226 bovinního PrP (tab. 1). Jedná se tedy o epitop složený, který podle autorů vzniká buď asociací dvou nebo více molekul prionového proteinu, nebo strukturálními změnami v rámci jedné molekuly PrP [38]. Protilátka byla patentována, nicméně se od té doby neobjevila ani na trhu s diagnostickými testy, ani v jiné vědecké práci zaměřené na použití v diagnostice. Až v roce 2008 Biasini et al ve studii na mutantních formách PrP konformačně specifickými protilátkami uveřejnili, že protilátka 15B3 má mnohem širší reaktivitu, než se původně myslelo [50]. Vedle proteáza rezistentních i senzitivních forem PrPTSE z infikovaných vzorků je 15B3 schopna imunoprecipitovat i neinfekční prionové agregáty, které se spontánně tvoří u neinfikovaných transgenních myší s nadprodukcí PrP a s inserčními, delečními nebo substitučními mutacemi genu pro PrP. Autoři ve své studii šli ještě dále a zjistili, že 15B3 je schopna detekovat i uměle precipitovaný PrPC z mozkových homogenátů zdravých „wild type“ myší nebo agregovanou formu rekombinantního (neinfekčního) PrP [50].

Hybridní IgG protilátky
V roce 2004 Moroncini et al připravili dvě hybridní protilátky obsahující peptidy PrPC schopné specificky vázat PrPTSE [51]. Autoři nejprve identifikovali sekvence, kterými se PrPC váže na molekulu PrPTSE [52]. Vazebné sekvence 89–112 a 136–158 myšího PrPC pak vložili do recipientní protilátky IgG1 12b [53] v oblasti determinující komplementaritu na těžkém řetězci. Takto připravené rekombinantní protilátky IgG 89–112 a IgG 136–158 imunoprecipitovaly myší, lidský a křeččí PrPTSE, ale ne rozpustný PrPC [51]. O několik let později se stejná laboratoř pokusila zmapovat vazebná místa PrPC na PrPTSE přípravou hybridních protilátek obsahujících postupně peptidy celé sekvence myšího prionového proteinu (moPrP23–231) [54]. Z 20 připravených protilátek s PrPTSE specificky reagovaly pouze tři: nově IgG 19–33 a IgG 89–112 a 136–158 identifikované již v roce 2004 Moroncinim et al (tab. 1). Všechny tři konformačně specifické hybridní protilátky reagovaly, stejně jako 15B3, i s agregáty neinfekčního PrPC a s agregáty neinfekčního rekombinantního PrP [50].
Tyr-Tyr-Arg protilátky
Další konformačně specifické protilátky vznikly imunizací peptidem obsahujícím aminokyselinový motiv Tyr-Tyr-Arg (Glu), který se nachází na třech místech molekuly PrP a je u savců mezidruhově konzervativní (tab. 1). Tyrosinové zbytky motivu Tyr-Tyr-Arg jsou v molekule PrPC orientovány dovnitř, zatímco u PrPTSE jsou exponovány na povrch [43] a představují PrPTSE-specifický epitop. Získané protilátky selektivně imunoprecipitují PrPTSE z infikovaných myších, křeččích, ovčích, bovinních a lidských mozkových homogenátů, avšak vedle PrPTSE rozeznávají i nefyziologicky složený a proteáza-senzitivní prionový protein [43]. Protilátky se na trhu s diagnostickými testy neobjevily, nicméně motiv konformačně specifického epitopu Tyr-Tyr-Arg využila v roce 2009 skupina S. Nappera, která se pokusila vyvinout vakcínu proti prionovým chorobám. Autoři nejprve optimalizovali epitop ovčího prionového proteinu obsahujícího motiv Tyr-Tyr-Arg tak, aby došlo k dostatečně silné imunitní odpovědi a zároveň nedošlo k aktivaci imunitního systému vůči PrPC. Po ukončení imunizace byla u ovcí zaznamenána PrPTSE specifická imunitní odpověď, specifické protilátky třídy IgG byly stanoveny v séru, mozkomíšním moku a mukózních sekretech a žádná z imunizovaných ovcí nevykazovala autoimunitní reakci proti PrPC [44]. Nicméně účinnost této vakcíny v prevenci a léčbě prionových chorob nebyla zatím prokázána.
V5B2
Imunizaci konformačně specifickými peptidy využila i skupina profesorky Šerbecové k přípravě protilátky V5B2 [55]. Ze studií zaměřených na identifikaci PrPTSE--specifických epitopů vybrali tři peptidy ze sekvence lidského PrP (P1: 214–226, P2: 167–179 a P3: 139–150), které použili k imunizaci BALB/c myší. Z připravených protilátek třídy IgG nejlépe reagovala protilátka V5B2 namířená proti C terminální části lidského PrP (sekvence 214–226) (tab. 1). V5B2 se zpočátku zdála reagovat PrPTSE specificky: rozeznávala CJN+ mozkové homogenáty detekované dot blotem nebo ELISOU od CJN-vzorků, se kterými nereagovala. Na rozdíl od většiny ostatních konformačně specifických protilátek nereagovala V5B2 s agregovaným ani s rozpustným rekombinantním lidským nebo bovinním PrP (23–230), reagovala pouze s peptidem P1 (214–226) [55]. Další několikaletou prací s protilátkou V5B2 však bylo zjištěno, že protilátka nerozeznává PrPTSE od PrPC na základě konformačních rozdílů, ale pouze rozeznává fragment molekuly PrPC zakončený aminokyselinou 226 [56].
OCD4 a g5p
V roce 2004 dokázali Zou et al specificky rozlišit PrPTSE od PrPC pomocí anti-DNA protilátek a genového proteinu 5 (g5p). Ke studii použili monoklonální protilátku OCD4 namířenou proti jaderné DNA a g5p používaný k detekci jednořetězcové DNA [57]. OCD4 a g5p rozlišovaly komplex DNA-PrPTSE, který se u PrPC nevyskytuje. Studie interakcí rekombinantního PrP s nukleovými kyselinami ukázaly, že DNA je schopna konvertovat α helikální strukturu C terminální části prionového proteinu do β skládaného listu [58]. Podle autorů studie dochází v této části molekuly vlivem konformačních změn prionového proteinu ke tvorbě stabilního komplexu DNA-PrPTSE a vazba není zrušena ani působením nukleázy (benzonázy), ani působením proteinázy K [57]. OCD4 a g5p specificky imunoprecipitovaly PrPTSE z mozkového homogenátu případů s CJN, zatímco s negativními homogenáty ani s homogenáty z jiných neurologických případů (např. AD) nereagovaly. Protilátka OCD4 byla schopná detekovat jak různé kmeny lidských prionů (sporadickou, dědičnou i získanou formu CJN a případy GSS), tak i různé zvířecí prionové kmeny (bovinní, ovčí, jelení, křeččí a myší). Zhruba 20 % PrPTSE imunoprecipitovaného pomocí OCD4 bylo proteáza-senzitivní [57].
P1:1
Jones et al použili k přípravě protilátek agregovaný peptid 106–126 lidského PrP [47]. Jedná se o centrální část prionového proteinu, která je u savců mezidruhově vysoce konzervovaná (tab. 1) a která během konformačních změn mění svoji strukturu z neuspořádané na strukturu β skládaného listu. Imunizací Prnp0/0 myší agregovaným peptidem 106–126 sekvence lidského PrP byla získána monoklonální protilátka P1:1 třídy IgM, která v nativních podmínkách selektivně imunoprecipitovala PrPTSE z lidských tkání. S negativními kontrolami ani s tkáněmi pacientů s jinými neurologickými diagnózami P1:1 nereagovala. Protilátka byla navíc schopna rozlišovat mezi typem 1 a 2 lidských prionových onemocnění charakterizovaných rozdílnou elektroforetickou mobilitou PrPres [59]. Autoři nicméně neprovedli testy reaktivity P1:1 s agregovaným rekombinantním (neinfekčním) prionovým proteinem ani s uměle agregovaným neinfekčním PrPC v homogenátech zdravých mozků.
6H10
Horiuchi et al připravili monoklonální protilátky třídy IgG imunizací Prnp0/0 myší nedenaturovaným PrPTSE, purifikovaným z infikovaného myšího mozkového homogenátu. Jedna z protilátek, 6H10, reagovala s nedenaturovaným myším PrPTSE, ale ne s rekombinantním myším PrP ani s denaturovaným PrPTSE [48]. 6H10 specificky imunoprecipitovala PrPTSE z myších, ovčích a bovinních mozkových homogenátů před štěpením proteinázou K i po něm, zatímco s neinfikovanými homogenáty nereagovala. V reakci na Biasiniho studii otestovali autoři protilátku 6H10 i s agregovaným rekombinantním prionovým proteinem a jeho C terminálním fragmentem (PrP23–231, resp. PrP89–231), se kterými protilátka nereagovala. 6H10 reagovala s mozkovou tkání infikovaných myší na histoblotu před štěpením proteinázou K i po něm. Reaktivita 6H10 byla výrazně redukována autoklávováním histoblotu, během kterého došlo pravděpodobně ke zrušení PrPTSE specifického epitopu. Podobně reaktivita 6H10 postupně slábla se zvyšující se koncentrací denaturačního činidla guanidin hydrochloridu. Při snaze o bližší charakterizaci vazebného epitopu autoři prokázali, že se jedná o složený, PrPTSE-konformačně specifický epitop (tab. 1), jehož částí jsou některé aminokyseliny C konce molekuly PrP (sekvence 215–228).
W261
Purifikovaný PrPTSE k přípravě konformačně specifických protilátek použili i Petsch et al v roce 2011 [49]. Autoři izolovali PrPTSE z infikovaných myších mozkových homogenátů vysrážením s kyselinou fosforečno-wolframovou a použili ho k imunizaci Prnp0/0 myší. Jedna z připravených protilátek, W261, třídy IgG, selektivně imunoprecipitovala PrPTSE z myších, křeččích, ovčích, jeleních a lidských mozkových homogenátů, zatímco se vzorky tkání zdravých jedinců nereagovala. Izolovaný PrPTSE byl částečně odolný vůči štěpení proteinázou K. Autoři ve snaze ukázat použitelnost protilátky W261 k diagnostickému testování zavedli sendvičovou ELISU, kde jako vyvazující protilátku použili W261 a k detekci běžně používanou anti-PrP protilátku 6H4 konjugovanou s peroxidázou. Následnou chemiluminiscencí byl bez použití proteinázy K detekován prionový protein z infikovaných ovčích (scrapie) a lidských (variantní CJN) mozkových homogenátů, ale ne ze zdravých kontrolních vzorků. Autoři však nevylučují, že by W261 mohla vázat i neinfekční agregáty prionového proteinu [49].
PRIOC
Další skupina se pokusila připravit PrPTSE specifické protilátky imunizací Prnp0/0 myší infikovaným myším mozkovým homogenátem štěpeným proteinázou K a adsorbovaným na magnetické mikropartikule (Dynabeads). Byly tak připraveny monoklonální protilátky PRIOC 1–4, třídy IgM, schopné detekovat rozpustné oligomery PrPTSE a dalších amyloidogenních proteinů, ale ne jejich monomery [60]. Při bližším určování epitopu nebyly protilátky schopny vázat rekombinantní ani buněčný PrP, ale byly schopny vázat kratší syntetické peptidy. PRIOC 1 a 2 rozeznávaly sekvenci PrP90–109 a PRIOC 3 a 4 sekvenci PrP170–189 (tab. 1). Protilátky selektivně vyvazovaly nativní (nedenaturovaný) PrPTSE z myších (RML) a lidských (vCJN) mozkových homogenátů před proteolýzou i po ní, zatímco se zdravými kontrolami nereagovaly. Žádná z protilátek překvapivě nereagovala s tkáněmi případů sporadické CJN. Autoři dále vyvinuli sendvičovou ELISU k detekci oligomerů PrPTSE v nedenaturujících podmínkách. PRIOC protilátku ukotvili na stěnu destičky a po vyvázání antigenu z RML infikovaného mozkového homogenátu použili tutéž protilátku k detekci v biotinylované formě. Všechny čtyři protilátky dobře detekovaly oligomery PrPTSE, ale s monomery PrP nedávaly žádný signál. Stejný systém byl použit i k testování rekombinantního prionového proteinu, protilátky detekovaly pouze rozpustné oligomery recPrP, s monomery ani s nerozpustnými fibrilami recPrP nereagovaly. Podobných výsledků bylo za použití PRIOC protilátek dosaženo i při detekci oligomerů peptidu amyloidu β (Aβ) a α synukleinu [60]. Protilátky schopné rozeznat rozpustné oligomery amyloidogenních proteinů byly připraveny i jinou skupinou v roce 2003. Kayed et al připravili oligomer-specifické polyklonální sérum imunizací králíků syntetickými oligomery peptidu Aβ [61]. Sérum nereagovalo s monomery ani s fibrilami amyloidu β, ale reagovalo s oligomery α synukleinu, amylinu (IAPP), polyglutaminu, lysozymu, lidského inzulinu a prionového peptidu 106–126. Autoři ve studii uvedli, že všechny výše zmíněné rozpustné oligomery sdílejí stejnou konformačně-dependentní strukturu, která je nezávislá na sekvenci [61].
Závěr
Použití konformačně specifických protilátek by vedle detekce proteáza-rezistentního PrPres umožnilo detekovat i proteáza-senzitivní formy PrPTSE, které se u některých případů prionových onemocnění zdají být jediným zdrojem patogeneze i diagnostické informace. Protilátek schopných rozeznat proteáza-senzitivní PrPTSE byla za posledních 15 let vyvinuta celá řada (tab. 2), nicméně otázkou zůstává jejich využití v praxi. Jejich užití pro detekci senPrPTSE v mozkové tkáni při post mortem diagnostice prionových chorob je limitováno nutností použití nedenaturujících technik zachovávajících konformaci senPrPTSE. Bohužel tyto techniky, např. imunoprecipitace, jsou často méně robustní a mohou vést k falešným výsledkům. Vše navíc komplikují vlastnosti buněčného prionového proteinu, PrPC, a jeho ochota tvořit po solubilizaci membrán agregáty v nedenaturujícím prostředí. U většiny konformačních protilátek proti PrPTSE bylo prokázáno, že vážou také agregovaný neinfekční PrP, a tudíž mohou poskytnout falešně pozitivní výsledky. Zdá se tedy, že širšímu použití konformačně specifických protilátek v diagnostice prionových chorob stojí v cestě především vývoj odpovídající detekční techniky. Za nadějné lze považovat v publikacích popsané ELISA testy [51,61], ale i ty čeká důsledná validace s klinickými vzorky. Za v současné době nejpropracovanější metodu schopnou detekce senPrPTSE lze považovat „Conformation Dependent Immunoassay – CDI“ vyvinutý Jiřím Šafářem et al [23,24]. Metoda CDI je založena na opačném přístupu, využívá protilátku 3F4, která rozeznává PrPC, ale v molekule PrPTSE je její epitop skryt. Epitop v molekule PrPTSE lze odkrýt denaturací vzorku. Rozdíl signálu mezi denaturovaným a nativním vzorkem pak odpovídá množství PrPTSE včetně senPrPTSE ve vzorku. Pro vzorky normálních tkání je tento rozdíl nulový. Alternativou ke konformačně specifickým testům by mohlo být nalezení kovalentní modifikace prionového proteinu, která by byla specifická pro PrPTSE a u PrPC by se nevyskytovala. Kandidátem takové modifikace je glykace, neenzymatická reakce redukujících cukrů s volnými aminoskupinami proteinů, jež pro svou pomalou rychlost může preferenčně modifikovat stabilní amyloidová depozita PrPTSE, zatímco PrPC zůstává pro svůj krátký buněčný poločas neglykovaný [62,63]. Protilátky specifické pro glykovaný PrPTSE by pak mohly být použitelné pro jeho detekci i za denaturujících podmínek [64], např. pomocí western blotu. Obrovským pokrokem, který by mohly přinést konformačně specifické protilátky, by byla rutinní detekce senPrPTSE v krvi nebo mozkomíšním moku pacientů umožňující potvrzení diagnózy před smrtí pacienta. Tyto tkáně zřejmě obsahují jen nepatrné množství PrPres, avšak obsah senPrPTSE by v nich mohl být vyšší [26]. Včasná diagnóza je nezbytným předpokladem pro vývoj a posléze i implementaci dnes neexistující účinné terapie. V současnosti se v diagnostice prionových chorob začínají uplatňovat vysoce rozlišující zobrazovací metody, jako jsou různé formy magnetické rezonance [65,66], avšak ani ty se neobejdou bez potvrzení nálezu konfirmačním testem. Další oblastí, kde by spolehlivý pre mortem test měl hrát důležitou úlohu, je prevence možného nozokomiálního šíření prionových chorob v rámci invazivních lékařských zákroků, například v neurochirurgii. V naší republice jsou na přítomnost PrPTSE povinně vyšetřováni všichni dárci oční rohovky, všechny vyšetřené vzorky byly zatím negativní [67]. Na druhou stranu zavedení univerzálního testování dárců krve se nezdá být ve většině států světa za současné situace praktické. Epidemiologická data nenaznačují, že by transfuze krve hrála důležitou úlohu v přenosu klasické CJN a počet úmrtí na vCJN ve Velké Británii se od roku 2005 drží na pěti a méně případech ročně. Navíc přes prokázanou přenositelnost vCJN krevní transfuzí [7] zatím nedochází k nárůstu počtu takto způsobených případů. Kromě vysokých nákladů by univerzální testování přineslo i nemalé etické problémy spojené s informováním zdravého dárce o možnosti, že má neléčitelnou neurodegenerativní chorobu. Nicméně to v žádném případě neznamená, že by se vědecká komunita neměla snažit o co nejrychlejší vývoj preklinického testu pro prionová onemocnění. Probíhající epidemie chronického chřadnutí (CWD) jelenovité zvěře v Severní Americe a obavy z možného přenosu CWD na hospodářská zvířata a člověka naznačují, že s prionovými chorobami je nutné počítat i do budoucna.
Samostatnou kapitolu tvoří otázka možného terapeutického použití PrPTSE specifických protilátek. Řada protilátek proti různým epitopům PrPC byla schopna inhibovat propagaci prionů in vitro a vyléčit priony infikované buněčné kultury [68]. Některé z protilátek byly navíc schopné prodloužit dobu přežití experimentálně infikovaných zvířat, zejména pokud byly podány profylakticky [69]. Předpokládá se, že vazba protilátky na PrPC ovlivňuje dynamiku jeho interakce s PrPTSE. Tím dojde k zpomalení propagace PrPTSE a buňka získá čas na jeho odbourání. Terapeutické použití protilátek proti PrPC je však spojeno s nebezpečím, že vyvázání buněčného PrPC povede vzhledem k jeho nejasné fyziologické úloze k nepředvídatelné a potenciálně nebezpečné reakci organizmu. Intracerebrální aplikace PrPC protilátek, ale i jejich F(ab)2 a Fab fragmentů vedla u transgenních myší k neurodegeneraci [70]. Toto nebezpečí by mohlo pomoci překlenout právě použití PrPTSE specifických protilátek, které se na normální PrPC v těle neváží. Samozřejmě i v tomto případě by komplikujícím faktorem terapie zůstala nutnost překonání hematoencefalické bariéry. Přes tyto problémy zůstává vývoj imunoterapie pro prionové choroby, ale i pro ostatní neurodegenerativní proteinopatie velmi nadějnou a atraktivní možností [71].
Závěrem lze shrnout, že konformačně specifické protilátky detekující senPrPTSE představují perspektivní nástroj, který by měl pomoci nejen s diagnostikou proteáza-senzitivní prionopatií, ale i s detekcí prionů u preklinických případů CJN, a umožnit tak prevenci nozokomiálního přenosu prionových chorob a zároveň získat čas pro aplikaci dnes chybějící účinné terapie.
Zdroj podpory: grant IGA MZ NS10335-3
Autoři děkují dr. Olze Janouškové (1. LF UK v Praze) za poskytnutí obrázku western blotu.
Ing. Karel Holada, Ph.D.
Ústav imunologie a mikrobiologie
1. LF UK v Praze
Studničkova 7
128 00 Praha 2
e-mail: Karel.Holada@lf1.cuni.cz
Přijato k recenzi: 4. 10. 2011
Přijato do tisku: 28. 11. 2011
Zdroje
1. Prusiner SB. Prions. Proc Natl Acad Sci U S A 1998; 95(23): 13363–13383.
2. Creutzfeldt HG. Über eine eigenartige herdförmige Erkrankung des Zentralnervensystems. Vorläufige Mitteilung. Z ges Neurol und Psychiat 1920; 57: 1–18.
3. Jakob A. Über eigenartige Erkrankungen des Zentralnervensystems mit bemerkenswerten anatomischen Befunde (spastische Pseudosklerose-Encephalomyelopathie mit disseminierten Degenerationsherden). Vorläufige Mitteilung. Deutsche Zeitschrift für Nervenheilkunde 1921; 70: 132–146.
4. Gajdusek DC, Zigas V. Degenerative disease of the central nervous system in New Guinea; the endemic occurrence of kuru in the native population. N Engl J Med 1957; 257(20): 974–978.
5. Matěj R, Rusina R, Koukolík F. 5 let činnosti Národní referenční laboratoře lidských prionových onemocnění při Oddělení patologie a molekulární medicíny FTNsP: naše zkušenosti a přehled literatury. Cesk Slov Neurol N 2007; 70/103(6): 637–642.
6. Will RG, Ironside JW, Zeidler M, Cousens SN, Estibeiro K, Alperovitch A et al. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet 1996; 347(9006): 921–925.
7. Hewitt PE, Llewelyn CA, Mackenzie J, Will RG. Creutzfeldt-Jakob disease and blood transfusion: results of the UK Transfusion Medicine Epidemiological Review study. Vox Sang 2006; 91(3): 221–230.
8. Holada K, Simak J, Brown P, Vostal JG. Divergent Expression of Cellular Prion Protein (PrPC) on Blood Cells of Human and Non-human Primates. Transfusion 2007; 47(12): 2223–2232.
9. Aguzzi A, Baumann F, Bremer J. The prion’s elusive reason for being. Annu Rev Neurosci 2008; 31: 439–477.
10. Borchelt DR, Scott M, Taraboulos A, Stahl N, Prusiner SB. Scrapie and cellular prion proteins differ in their kinetics of synthesis and topology in cultured cells. J Cell Biol 1990; 110(3): 743–752.
11. Caughey B, Race RE, Ernst D, Buchmeier MJ, Chesebro B. Prion protein biosynthesis in scrapie-infected and uninfected neuroblastoma cells. J Virol 1989; 63(1): 175–181.
12. Pan KM, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D et al. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci U S A 1993; 90(23): 10962–10966.
13. Safar J, Roller PP, Gajdusek DC, Gibbs CJ jr. Conformational transitions, dissociation, and unfolding of scrapie amyloid (prion) protein. J Biol Chem 1993; 268(27): 20276–20284.
14. Govaerts C, Wille H, Prusiner SB, Cohen FE. Evidence for assembly of prions with left-handed beta--helices into trimers. Proc Natl Acad Sci U S A 2004; 101(22): 8342–8347.
15. Horiuchi M, Caughey B. Specific binding of normal prion protein to the scrapie form via a localized domain initiates its conversion to the protease-resistant state. EMBO J 1999; 18(12): 3193–3203.
16. Hobzová K, Janoušková O. Tkáňové kultury pro studium prionových chorob. Cesk Slov Neurol N 2010; 73(4): 379–386.
17. Collinge J, Gorham M, Hudson F, Kennedy A, Keogh G, Pal S et al. Safety and efficacy of quinacrine in human prion disease (PRION-1 study): a patient-preference trial. Lancet Neurol 2009; 8(4): 334–344.
18. Tsuboi Y, Doh-Ura K, Yamada T. Continuous intraventricular infusion of pentosan polysulfate: clinical trial against prion diseases. Neuropathology 2009; 29(5): 632–636.
19. Zerr I. Therapeutic trials in human transmissible spongiform encephalo-pathies: recent advances and problems to address. Infect Disord Drug Targets 2009; 9(1): 92–99.
20. Glierová H, Holada K. Úloha imunitního systému v prionových chorobách. Alergie 2006; 8(2): 143–148.
21. Sassa Y, Kataoka N, Inoshima Y, Ishiguro N. Anti-PrP antibodies detected at terminal stage of prion-affected mouse. Cell Immunol 2010; 263(2): 212–218.
22. Holada K. Prionové choroby a pokrok ve vývoji testu pro preklinickou detekci abnormálního prionového proteinu (PrPsc). Klin Mikrobiol Inf Lék 2005; 11(5): 151–160.
23. Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M et al. Eight prion strains have PrP(Sc) molecules with different conformations. Nat Med 1998; 4(10): 1157–1165.
24. Safar JG, Geschwind MD, Deering C, Didorenko S, Sattavat M, Sanchez H et al. Diagnosis of human prion disease. Proc Natl Acad Sci U S A 2005; 102(9): 3501–3506.
25. Thackray AM, Hopkins L, Bujdoso R. Proteinase K-sensitive disease-associated ovine prion protein revealed by conformation-dependent immunoassay. Biochem J 2007; 401(2): 475–483.
26. Yakovleva O, Janiak A, McKenzie C, McShane L, Brown P, Cervenakova L. Effect of protease treatment on plasma infectivity in variant Creutzfeldt-Jakob disease mice. Transfusion 2004; 44(12): 1700–1705.
27. Holada K, Glierova H, Simak J, Vostal JG. Expression of cellular prion protein on platelets from patients with gray platelet or Hermansky-Pudlak syndrome and the protein‘s association with alpha-granules. Haematologica 2006; 91(8): 1126–1129.
28. Holada K, Simak J, Vostal JG. Transmission of BSE by blood transfusion. Lancet 2000; 356(9243): 1772.
29. Holada K, Simak J, Risitano AM, Maciejewski J, Young NS, Vostal JG. Activated platelets of patients with paroxysmal nocturnal hemoglobinuria express cellular prion protein. Blood 2002; 100(1): 341–343.
30. Daude N, Lehmann S, Harris DA. Identification of intermediate steps in the conversion of a mutant prion protein to a scrapie-like form in cultured cells. J Biol Chem 1997; 272(17): 11604–11612.
31. Horiuchi M, Priola SA, Chabry J, Caughey B. Interactions between heterologous forms of prion protein: binding, inhibition of conversion, and species barriers. Proc Natl Acad Sci U S A 2000; 97(11): 5836–5841.
32. Pastrana MA, Sajnani G, Onisko B, Castilla J, Morales R, Soto C et al. Isolation and characterization of a proteinase K-sensitive PrPSc fraction. Biochemistry 2006; 45(51): 15710–15717.
33. Gambetti P, Dong Z, Yuan J, Xiao X, Zheng M, Alshekhlee A et al. A novel human disease with abnormal prion protein sensitive to protease. Ann Neurol 2008; 63(6): 697–708.
34. Jansen C, Parchi P, Jelles B, Gouw AA, Beunders G, van Spaendonk RM et al. The first case of fatal familial insomnia (FFI) in the Netherlands: a patient from Egyptian descent with concurrent four repeat tau deposits. Neuropathol Appl Neurobiol 2011; 37(5): 549–553.
35. Rodríguez-Martínez AB, Garrido JM, Zarranz JJ, Arteagoitia JM, de Pancorbo MM, Atarés B et al. A novel form of human disease with a protease-sensitive prion protein and heterozygosity methionine//valine at codon 129: Case report. BMC Neurol 2010; 10: 99.
36. Head MW, Knight R, Zeidler M, Yull H, Barlow A, Ironside JW. A case of protease sensitive prionopathy in a patient in the UK. Neuropathol Appl Neurobiol 2009; 35(6): 628–632.
37. Zou WQ, Puoti G, Xiao X, Yuan J, Qing L, Cali I et al. Variably protease-sensitive prionopathy: a new sporadic disease of the prion protein. Ann Neurol 2010; 68(2): 162–172.
38. Korth C, Stierli B, Streit P, Moser M, Schaller O, Fischer R et al. Prion (PrPSc)-specific epitope defined by a monoclonal antibody. Nature 1997; 390(6655): 74–77.
39. Spinner DS, Kascsak RB, Lafauci G, Meeker HC, Ye X, Flory MJ et al. CpG oligodeoxynucleotide-enhanced humoral immune response and production of antibodies to prion protein PrPSc in mice immunized with 139A scrapie-associated fibrils. J Leukoc Biol 2007; 81(6): 1374–1385.
40. Bainbridge J, Jones N, Walker B. Multiple antigenic peptides facilitate generation of anti-prion antibodies. Clin Exp Immunol 2004; 137(2): 298–304.
41. Büeler H, Fischer M, Lang Y, Bluethmann H, Lipp HP, DeArmond SJ et al. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 1992; 356(6370): 577–582.
42. Manson JC, Clarke AR, Hooper ML, Aitchison L, McConnell I, Hope J. 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol Neurobiol 1994; 8(2–3): 121–127.
43. Paramithiotis E, Pinard M, Lawton T, LaBoissiere S, Leathers VL, Zou WQ et al. A prion protein epitope selective for the pathologically misfolded conformation. Nat Med 2003; 9(7): 893–899.
44. Hedlin PD, Cashman NR, Li L, Gupta J, Babiuk LA, Potter A et al. Design and delivery of a cryptic PrP(C) epitope for induction of PrP(Sc)-specific antibody responses. Vaccine 2010; 28(4): 981–988.
45. Thackray AM, Madec JY, Wong E, Morgan-Warren R, Brown DR, Baron T et al. Detection of bovine spongiform encephalopathy, ovine scrapie prion-related protein (PrPSc) and normal PrPC by monoclonal antibodies raised to copper-refolded prion protein. Biochem J 2003; 370(1): 81–90.
46. Polymenidou M, Moos R, Scott M, Sigurdson C, Shi YZ, Yajima B et al. The POM monoclonals: a comprehensive set of antibodies to non-overlapping prion protein epitopes. PLoS One 2008; 3(12): e3872.
47. Jones M, Wight D, McLoughlin V, Norrby K, Ironside JW, Connolly JG et al. An antibody to the aggregated synthetic prion protein peptide (PrP106–126) selectively recognizes disease-associated prion protein (PrP) from human brain specimens. Brain Pathol 2009; 19(2): 293–302.
48. Horiuchi M, Karino A, Furuoka H, Ishiguro N, Kimura K, Shinagawa M. Generation of monoclonal antibody that distinguishes PrPSc from PrPC and neutralizes prion infectivity. Virology 2009; 394(2): 200–207.
49. Petsch B, Müller-Schiffmann A, Lehle A, Zirdum E, Prikulis I, Kuhn F et al. Biological effects and use of PrPSc- and PrP-specific antibodies generated by immunization with purified full-length native mouse prions. J Virol 2011; 85(9): 4538–4546.
50. Biasini E, Seegulam ME, Patti BN, Solforosi L, Medrano AZ, Christensen HM et al. Non-infectious aggregates of the prion protein react with several PrPSc-directed antibodies. J Neurochem 2008; 105(6): 2190–2204.
51. Moroncini G, Kanu N, Solforosi L, Abalos G, Telling GC, Head M et al. Motif-grafted antibodies containing the replicative interface of cellular PrP are specific for PrPSc. Proc Natl Acad Sci U S A 2004; 101(28): 10404–10409.
52. Williamson RA, Peretz D, Pinilla C, Ball H, Bastidas RB, Rozenshteyn R et al. Mapping the prion protein using recombinant antibodies. J Virol 1998; 72(11): 9413–9418.
53. Burton DR, Pyati J, Koduri R, Sharp SJ, Thornton GB, Parren PW et al. Efficient neutralization of primary isolates of HIV-1 by a recombinant human monoclonal antibody. Science 1994; 266(5187): 1024–1027.
54. Solforosi L, Bellon A, Schaller M, Cruite JT, Abalos GC, Williamson RA. Toward molecular dissection of PrPC-PrPSc interactions. J Biol Chem 2007; 282(10): 7465–7471.
55. Curin Serbec V, Bresjanac M, Popovic M, Pretnar Hartman K, Galvani V, Rupreht R et al. Monoclonal antibody against a peptide of human prion protein discriminates between Creutzfeldt-Jacob‘s disease-affected and normal brain tissue. J Biol Chem 2004; 279(5): 3694–3698.
56. Kosmač M, Koren S, Giachin G, Stoilova T, Gennaro R, Legname G et al. Epitope mapping of a PrP(Sc)-specific monoclonal antibody: identification of a novel C-terminally truncated prion fragment. Mol Immunol 2011; 48(5): 746–750.
57. Zou WQ, Zheng J, Gray DM, Gambetti P, Chen SG. Antibody to DNA detects scrapie but not normal prion protein. Proc Natl Acad Sci U S A 2004; 101(5): 1380–1385.
58. Cordeiro Y, Machado F, Juliano L, Juliano MA, Brentani RR, Foguel D et al. DNA converts cellular prion protein into the beta-sheet conformation and inhibits prion peptide aggregation. J Biol Chem 2001; 276(52): 49400–49409.
59. Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 1999; 46(2): 224–233.
60. Tayebi M, Jones DR, Taylor WA, Stileman BF, Chapman C, Zhao D et al. PrP(Sc)-specific antibodies with the ability to immunodetect prion oligomers. PLoS One 2011; 6(5): e19998.
61. Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003; 300(5618): 486–489.
62. Choi YG, Kim JI, Jeon YC, Park SJ, Choi EK, Rubenstein R et al. Nonenzymatic glycation at the N terminus of pathogenic prion protein in transmissible spongiform encephalopathies. J Biol Chem 2004; 279(29): 30402–30409.
63. Panigaj M, Brouckova A, Glierova H, Dvorakova E, Simak J, Vostal JG et al. Underestimation of the expression of cellular prion protein on human red blood cells. Transfusion 2011; 51(5): 1012–1021.
64. Dvorakova E, Prouza M, Janouskova O, Panigaj M, Holada K. Development of monoclonal antibodies specific for glycated prion proteins. J Toxicol Environ Health A 2011; 74(22–24): 1469–1475.
65. Tschampa HJ, Zerr I, Urbach H. Radiological assessment of Creutzfeldt-Jakob disease. Eur Radiol 2007 May; 17(5): 1200–1211.
66. Sheardová K, Matěj R, Rektorová I. Hyperintenzivní léze reagující na steroidy u pacienta s Creutzfeldt‑Jakobovou nemocí. Cesk Slov Neurol N 2010; 73/106(1): 76–79.
67. Jirsova K, Krabcova I, Novakova J, Hnathova I, Koukolik F, Kubesova B et al. The assessment of pathogenic prions in the brains of eye tissue donors: 2-years experience in the Czech Republic. Cornea 2010; 29(9): 996–999.
68. Peretz D, Williamson RA, Kaneko K, Vergara J, Leclerc E, Schmitt-Ulms G et al. Antibodies inhibit prion propagation and clear cell cultures of prion infectivity. Nature 2001; 412(6848): 739–743.
69. White AR, Enever P, Tayebi M, Mushens R, Linehan J, Brandner S et al. Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature 2003; 422(6927): 80–83.
70. Lefebvre-Roque M, Kremmer E, Gilch S, Zou WQ, Féraudet C, Gilles CM et al. Toxic effects of intracerebral PrP antibody administration during the course of BSE infection in mice. Prion 2007; 1(3): 198–206.
71. Wisniewski T, Goñi F. Immunomodulation for prion and prion-related diseases. Expert Rev Vaccines 2010; 9(12): 1441–1452.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2012 Číslo 3
Nejčtenější v tomto čísle
- Neurosyfilis
- Operační léčba syndromu tarzálního tunelu
- Oboustranná léze n. phrenicus manifestující se jako ortopnoe – kazuistiky tří případů
- Diagnostika a možnosti léčby Niemann-Pickovy choroby typ C