
Hereditární časná forma Alzheimerovy nemoci v důsledku zárodečné mutace p.M139V v genu PSEN1 – kazuistika
Early-Onset Hereditary Alzheimer’s Disease Caused by p.M139V Mutation in the PSEN1 Gene – a Case ReportAlzheimerova demence je nejčastější demence u pacientů ve starším věku. V některých rodinách může být geneticky podmíněna. Naše kazuistika ukazuje případ 43letého muže, v jehož rodině se vyskytlo dalších šest členů rodiny s manifestací demence ve věku 40–50 let. Genetickým vyšetřením byla u pacienta prokázána patogenní mutace c.415A>G (p.M139V) v exonu 5 genu PSEN1 v heterozygotním stavu. Stejná mutace byla zjištěna u demencí postiženého bratrance. V rodině tak byla potvrzena hereditární predispozice k časné formě Alzheimerovy demence s autozomálně dominantní dědičností na molekulární úrovni. Vývoj onemocnění byl u pacienta sledován po dobu osmi let. Postupně dochází k deterioraci kognitivních funkcí a vývoji atrofických změn mozku dle magnetické rezonance. Obdobné změny jsou pozorovány u jeho bratrance. Genetické vyšetřování v rodinách zasažených demencí může být do budoucna důležité především pro možnost včasné léčby pacientů v riziku.
Alzheimer’s disease is the most frequently diagnosed form of dementia in older patients. In some families, a tendency to this disorder may be inherited. This case report describes a 43-year-old man in the family of whom are six other members suffering from dementia, between the ages of 40 and 50 years. Molecular genetic analysis revealed a pathogenic c.415A>G (p.M139V) mutation in exon 5 of the PSEN1 gene in the heterozygous state. The same mutation was found in a cousin who has also suffered from dementia since the age of 45. Thus a hereditary predisposition to the early-onset form of Alzheimer’s disease was confirmed in this family at a molecular level. The patient was followed up for eight years. Gradual deterioration of cognitive functions and progression of brain atrophy was observed on MRI. Similar changes were observed in the cousin. Genetic testing in families suffering from dementia may be important in the future, together with the development of drugs capable of preventing the disease.
Key words:
Alzheimer’s dementia – hereditary disease – the PSEN1 gene – gene mutation
Autoři:
P. Bártová 1; S. Walczysková 2; P. Plevová 2; L. Ratajová 3; J. Havelka 4; E. Šilhánová 2; P. Ressner 1; D. Školoudík 1,5
Působiště autorů:
Neurologická klinika OU a FN Ostrava
1; Oddělení lékařské genetiky, OU a FN Ostrava
2; Neurologická ambulance, Mladá Boleslav
3; Radiodiagnostické oddělení, OU a FN Ostrava
4; Neurologická klinika LF UP a FN Olomouc
5
Vyšlo v časopise:
Cesk Slov Neurol N 2011; 74/107(5): 569-574
Kategorie:
Kazuistika
Souhrn
Alzheimerova demence je nejčastější demence u pacientů ve starším věku. V některých rodinách může být geneticky podmíněna. Naše kazuistika ukazuje případ 43letého muže, v jehož rodině se vyskytlo dalších šest členů rodiny s manifestací demence ve věku 40–50 let. Genetickým vyšetřením byla u pacienta prokázána patogenní mutace c.415A>G (p.M139V) v exonu 5 genu PSEN1 v heterozygotním stavu. Stejná mutace byla zjištěna u demencí postiženého bratrance. V rodině tak byla potvrzena hereditární predispozice k časné formě Alzheimerovy demence s autozomálně dominantní dědičností na molekulární úrovni. Vývoj onemocnění byl u pacienta sledován po dobu osmi let. Postupně dochází k deterioraci kognitivních funkcí a vývoji atrofických změn mozku dle magnetické rezonance. Obdobné změny jsou pozorovány u jeho bratrance. Genetické vyšetřování v rodinách zasažených demencí může být do budoucna důležité především pro možnost včasné léčby pacientů v riziku.
Klíčová slova:
Alzheimerova demence – hereditární onemocnění – gen PSEN1 – genová mutace
Úvod
Demence postihuje přibližně 5,4 % osob ve věku nad 65 let a její prevalence narůstá s věkem [1]. V přibližně 5–10 % všech případů Alzheimerovy nemoci se jedná o familiární výskyt onemocnění [2]. Zatímco rizikovým faktorem pozdní formy onemocnění je varianta E4 v genu pro apolipoprotein E (chromozomální lokus 19q13.2), časná forma onemocnění s nástupem příznaků před 65. rokem věku může být způsobena patogenními mutacemi v jednom ze tří genů: v genech pro amyloidový prekurzorový protein (gen APP, chromozomální lokus 21q21), pro presenilin 1 (gen PSEN1, chromozomální lokus 14q24.3) nebo presenilin 2 (gen PSEN2, chromozomální lokus 1q31–42) [3]. V tomto případě se jedná o hereditární predispozici k časné formě onemocnění dědičnou autozomálně dominantně, kdy potomci nemocné osoby mají 50% pravděpodobnost, že vlohu k onemocnění zdědí. Většinu mutací v genech PSEN1 a APP charakterizuje manifestace onemocnění mezi 25. a 65. rokem života a úplná penetrance, kdy onemocní všechny geneticky predisponované osoby. Mutace v genu PSEN2 jsou spojeny s pozdějším věkem nástupu onemocnění (45 až 88 let) a penetrance je neúplná, kdy u některých osob, ačkoli genetickou vlohu k onemocnění nesou, se onemocnění nemusí vůbec klinicky manifestovat [4]. Tyto hereditární formy s autozomálně dominantním typem dědičnosti nejsou početné a v celé populaci nemocných tvoří sotva několik procent [5].
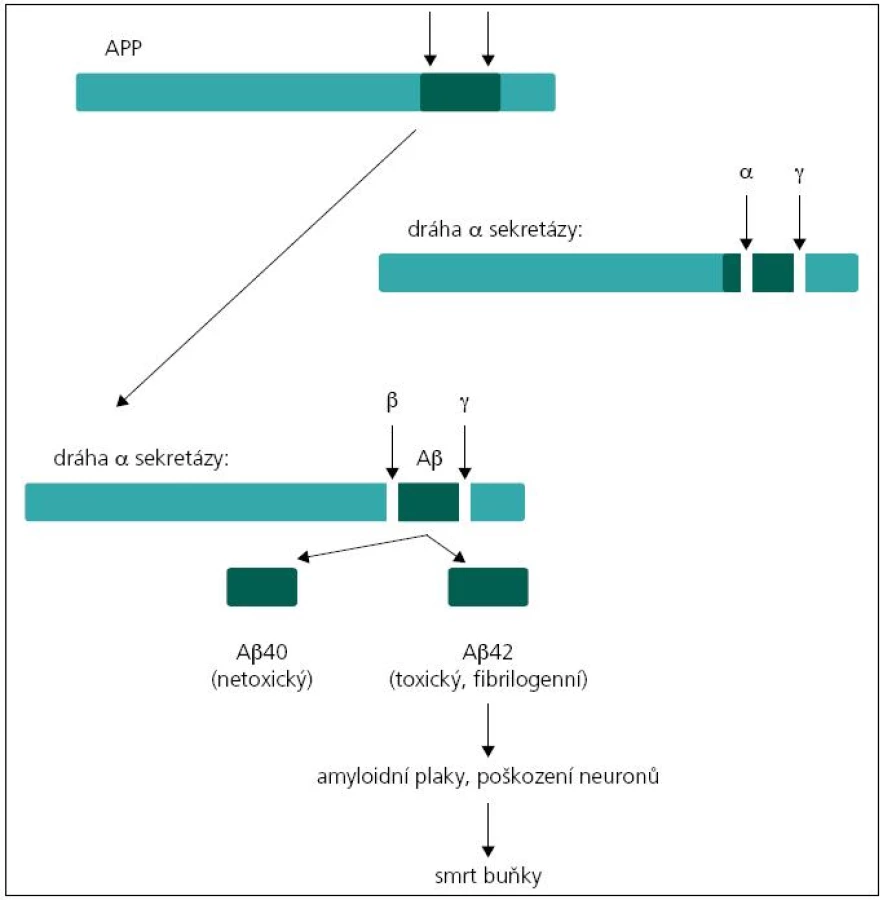
Existují dvě teorie týkající se patogeneze demence u pacientů s mutacemi v genech PSEN1, PSEN2 a APP. Podle první z nich, tzv. amyloidové hypotézy, je příčinou tvorba amyloidních plaků v mozkové tkáni. Hlavní komponentou amyloidních plaků je amyloid β (Aβ). Nejčastější forma amyloidu β u člověka má 40 aminokyselin (tzv. Aβ40). Méně častá forma je amyloid o 42 aminokyselinách, který se liší od předchozího pouze dvěma aminokyselinovými zbytky navíc (tzv. Aβ42), a právě ukládání této formy amyloidu v placích je spojeno s rozvojem Alzheimerovy choroby. Amyloid β vzniká štěpením transmembránového proteinu APP (amyloidový prekurzorový protein) sekretázami. Protein APP může být štěpen dvěma způsoby. Při prvním dojde k rozštěpení APP α-sekretázou, což je „standardní“ štěpení bez neurotoxického účinku. Při druhém probíhá štěpení APP nejprve β sekretázou a poté komplexem γ sekretázy. Proteinový komplex γ sekretázy je tvořen preseniliny 1 a 2 spolu s nikastrinem, PEN-2 a APH-1. Při tomto druhém způsobu štěpení mohou vznikat Aβ peptidy o variabilní délce řetězce 39 až 42 aminokyselin v závislosti na místě štěpení, nejčastěji však Aβ40 nebo Aβ42 (obr. 1). Podíl těchto forem je důležitý v patogenezi Alzheimerovy choroby, neboť Aβ42 má větší tendenci k agregaci, oligomerizaci a tvorbě fibril než Aβ peptidy o kratší délce [4,6,7]. V případě mutací v genech PSEN1 nebo PSEN2 dochází ke změně proteolytické aktivity komplexu γ sekretázy a selektivnímu zvýšení produkce amyloidu β, případně ke změně podílu kratších/delších forem Aβ peptidů [8,9]. Podle druhé, tzv. presenilinové hypotézy dochází v důsledku mutací genů ke ztrátě zásadních funkcí presenilinů, což vede přímo k neuronální degeneraci a ztrátě paměti bez tvorby amyloidních plaků [9].
V naší kazuistice popisujeme poprvé v České republice případ rodiny s hereditární predispozicí k časné formě Alzheimerovy choroby způsobenou patogenní mutací v genu PSEN1.
Popis případu
Pacient (obr. 2, IV-7) ve věku 43 let byl přiveden matkou k hospitalizaci na Neurologickou kliniku Fakultní nemocnice Ostrava v roce 2003 pro poruchy paměti a poruchy chování, jejichž pozvolný počátek byl patrný již od 37 let věku. Byla to především úzkostná porucha s prvky paranoidního chování, toulání, později i agresivita. Pacient netrpěl žádným jiným sledovaným onemocněním a žil ve společné domácnosti s matkou. Absolvoval pouze základní vzdělání. V té době ztratil i práci, protože selhával i v dělnických profesích – zapomínal posloupnost dějů a podobně.
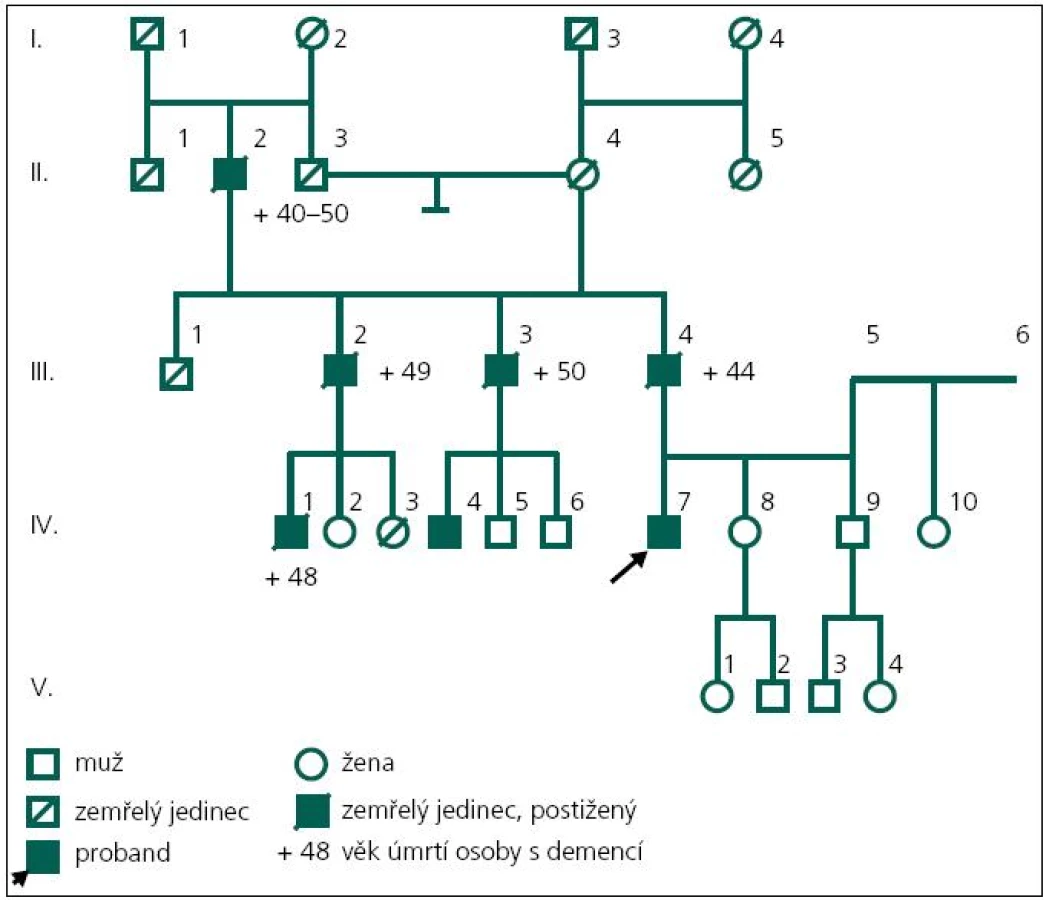
Matka pacienta zároveň poukazuje na určité podobné rysy v rodinné anamnéze; otec pacienta (obr. 2, III-4) byl opakovaně za svého života hospitalizován na psychiatrické klinice, byl agresivní, podlehl etylizmu a zemřel ve 44 letech na demenci. Provedený sekční nález z roku 1979 ukazoval atrofii mozku zejména frontálních laloků, hydrocefalus externus et internus e vacuo, především laterálních komor. Histologicky byl popsán úbytek korové hmoty, především vrstvy 1–3, úbytek neuronů i astroglií, atroficko-degenerativní změny pyramidových neuronů, změny odpovídající Alzheimerovým změnám neurofibril. Závěrečná diagnóza byla Alzheimerova demence.
Otec otce (obr. 2, II-2) také zemřel na demenci. Dva ze tří sourozenců otce (jeden muž a jedna žena; obr. 2, III-2 a III-3) byli také postiženi rozvojem demence a každý z nich má jednoho syna s manifestací demence ve 44 a 45 letech věku (obr. 2, IV-1, IV-4).
V objektivním neurologickém vyšetření v roce 2003 byly u pacienta popsány prvky mimovolných pohybů typu chorey, zvláště na horních končetinách, která se projevovala hlavně v sociálních kontaktech při anxietě. Při vyšetření Mini-Mental State Examination (MMSE) dosáhl pacient 20 bodů [10]. Toto vyšetření odhalilo hrubé poruchy orientace, pozornosti, počítání, výbavnosti, vizuospaciálních funkcí a písma. Psychologické vyšetření prokázalo oslabení intelektových schopností. Globální intelektový výkon odpovídal pásmu lehké mentální retardace – demenci. Byl zjištěn výrazný rozdíl mezi jednotlivými složkami, s dominancí verbálních složek nad performačními. Výrazně oslabena byla schopnost vštípivosti a retence z krátkodobých a dlouhodobých složek paměti. Hodnota MQ byla 64. Byly oslabeny symbolické funkce a celkově došlo k výraznému oslabení abstraktního myšlení a uvažování. Snížena byla také schopnost kognitivní flexe a organizace výbavnosti z dlouhodobých složek paměti. Výsledek vyšetření nasvědčuje exekutivnímu oslabení. S ohledem na zjištěné skutečnosti odpovídal aktuální stav stavu mírné až středně těžké demence se suspektním progredujícím charakterem a se sekundárním úzkostně depresivním prožíváním a agresivními projevy v emocionální zátěži.
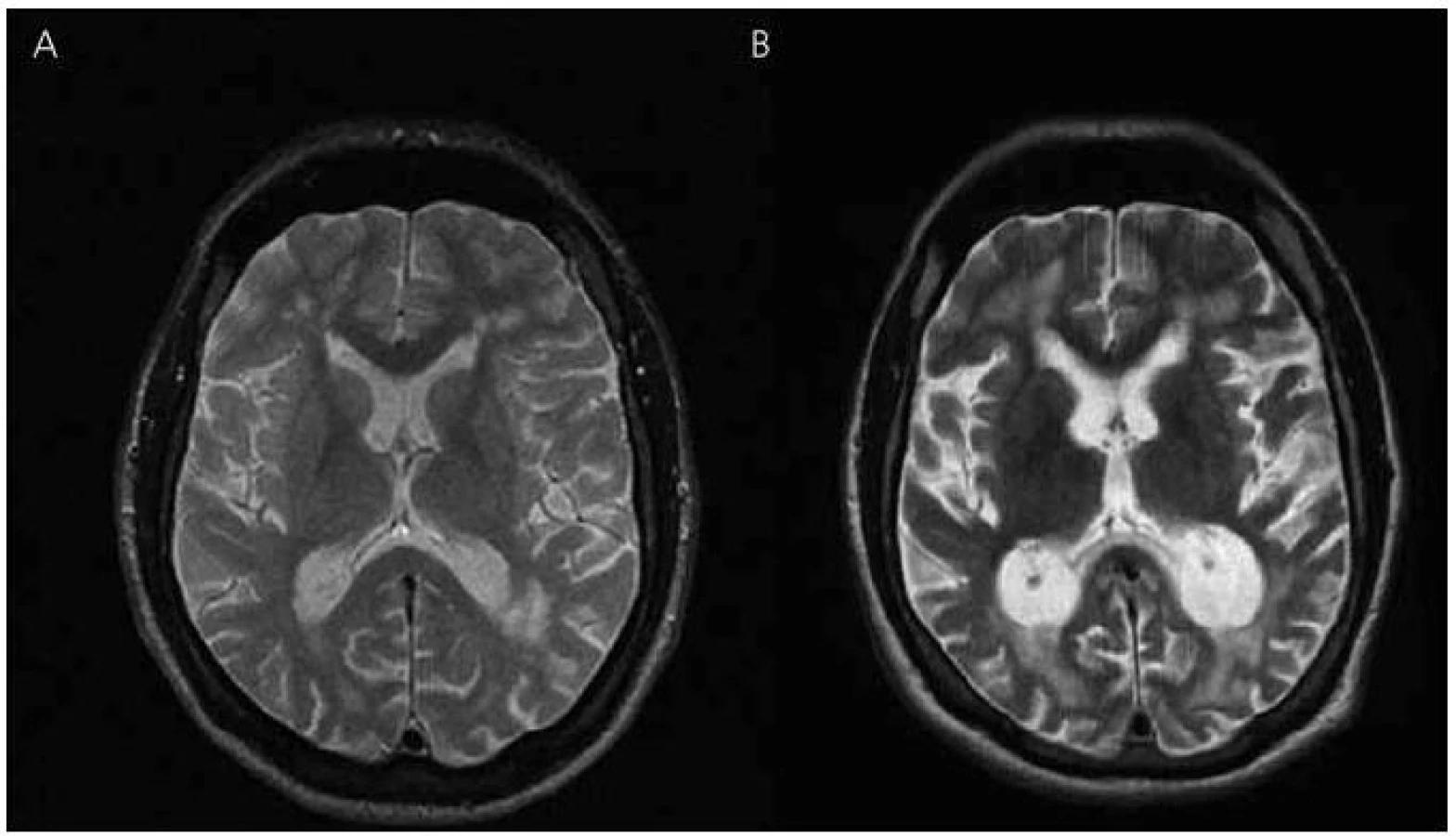
V roce 2003 bylo provedeno také první vyšetření mozku magnetickou rezonancí (MR), které ukázalo nevýrazná nespecifická ložiska demyelinizačního charakteru v bílé hmotě frontoparietálně oboustranně, bez známek atrofie mozku (obr. 3 a 4). Zároveň provedené elektroencefalografické vyšetření (EEG) ukázalo hrubě abnormní záznam pro nevyjádřenou hemisferální a areální organizaci, difuzní theta-beta aktivitu. Pacient se podrobil také vyšetření likvoru, kde však veškeré nálezy biochemické, cytologické, virologie, průkaz oligoklonálních pásů i protilátek proti boreliím byly negativní.

První genetické vyšetření bylo provedeno v témže roce. Vzhledem k choreatickým dyskinezám v úvodu byla u pacienta vyšetřována a zároveň vyloučena Westphalova forma Huntingtonovy choroby. Dále byla zvažována demence s autozomálně dominantním přenosem v rodině, a to frontotemporální demence (mutace genu MAPT 17q21), Alzheimerova nemoc (mutace genů APP, PSEN1 a PSEN2) a Creutzfeldtova-Jakobova nemoc (mutace genu PRNP), molekulárně genetické vyšetření těchto genů však nebylo v té době dostupné.
U pacienta byla stanovena diagnóza Alzheimerovy demence, dle diagnostických kritérií NINCDS-ADRDA [11,12]. Zároveň byla zahájena léčba inhibitory acetylcholinesterázy (rivastigmin do dávky 12 mg za den). Pacient začal na léčbě rivastigminem lépe spolupracovat s rodinou a začal se více zapojovat do aktivit v domácím prostředí. V testovém hodnocení kognitivních funkcí pacienta nedošlo ke zlepšení, pouze se zlepšila ochota komunikovat s lékařem.
Později byl nasazen pro neklid a agresivitu tiaprid 100 mg na noc. Matka byla poučena o nutnosti tréninku paměti s pacientem, který se snažila po celou dobu provádět.
V roce 2008 bylo indikováno druhé genetické vyšetření. Byla při něm indikována molekulárně genetická analýza genů pro časnou formu Alzheimerovy choroby PSEN1 a APP. DNA byla izolována z leukocytů periferní krve standardním postupem. Intronické primery byly použity k amplifikaci a sekvenaci exonů 3.–12. genu PSEN1 a exonů 16 a 17 genu APP [13]. Přímá sekvenace byla provedena na genetickém analyzátoru ABI3130. U vyšetřeného byla nalezena patogenní mutace c.415A>G v exonu 5 genu PSEN1 v heterozygotním stavu (tj. na jedné alele genu) (obr. 5). Jedná se o missense mutaci, tedy mutaci se změněným smyslem, která vede na úrovni proteinu k záměně původní aminokyseliny metioninu v kodónu 139 za valin (p.M139V). Tím byla u vyšetřeného potvrzena hereditární predispozice k časné formě Alzheimerovy choroby s autozomálně dominantním typem dědičnosti.

Zároveň byl nalezen genotyp APO-E*E2/4 v genu pro apolipoprotein E. Bylo zjištěno, že alelu E4 vyšetřený zdědil od své mentálně zcela zdravé matky, které je nyní 72 let (obr. 1, III-5; genotyp matky byl APOE*E3/4).
U pacienta dochází k postupné progresi demence i přes včasné nasazení léčby. MMSE klesá na 13 bodů, ale stále zůstává schopnost pacienta provádět aktivity v domácím prostředí s dopomocí rodiny, v jejíž péči pacient nadále setrvává. Po dvou letech průběhu choroby ustoupily dyskinetické pohyby horních končetin při sociálních kontaktech a začala převládat spíše apatie se zpomalenými psychomotorickými projevy. V této době byl přidán memantin, který však způsoboval výraznou žaludeční nevolnost u pacienta, proto byl vysazen. MR mozku v roce 2005 byla bez vývoje v porovnání s nálezem z roku 2003. Opakované vyšetření likvoru na hladiny specifických proteinů (tau proteinu, fosforylovaného tau proteinu a β-amyloidu) bylo v normě.
V roce 2010 se začíná objevovat apatie, progredují poruchy paměti, především anterográdní s poruchou vštípivosti a výbavnosti, porucha řeči s poklesem verbální fluence, agrafie a poruchy čtení textu. Zhoršuje se také fungování v aktivitách denního života. Pacient nezvládá posloupnost oblékání, nechce vycházet z domu, neumí používat příbor, objevují se poruchy příjmu potravy a občasné problémy s užíváním léků. Z důvodu nízké compliance při užívání léků byl přechodně převeden na rivastigmin náplasti 9,5 mg/den, které ale strhával. Proto byl převeden zpět na tabletovou formu. Nadále zůstává v péči matky. Kontrolní vyšetření MR v roce 2010 ukazuje rozvoj výrazné atrofie mozku, rozšíření komorového systému a subarachnoideálních prostorů konvexit (obr. 3, 4).
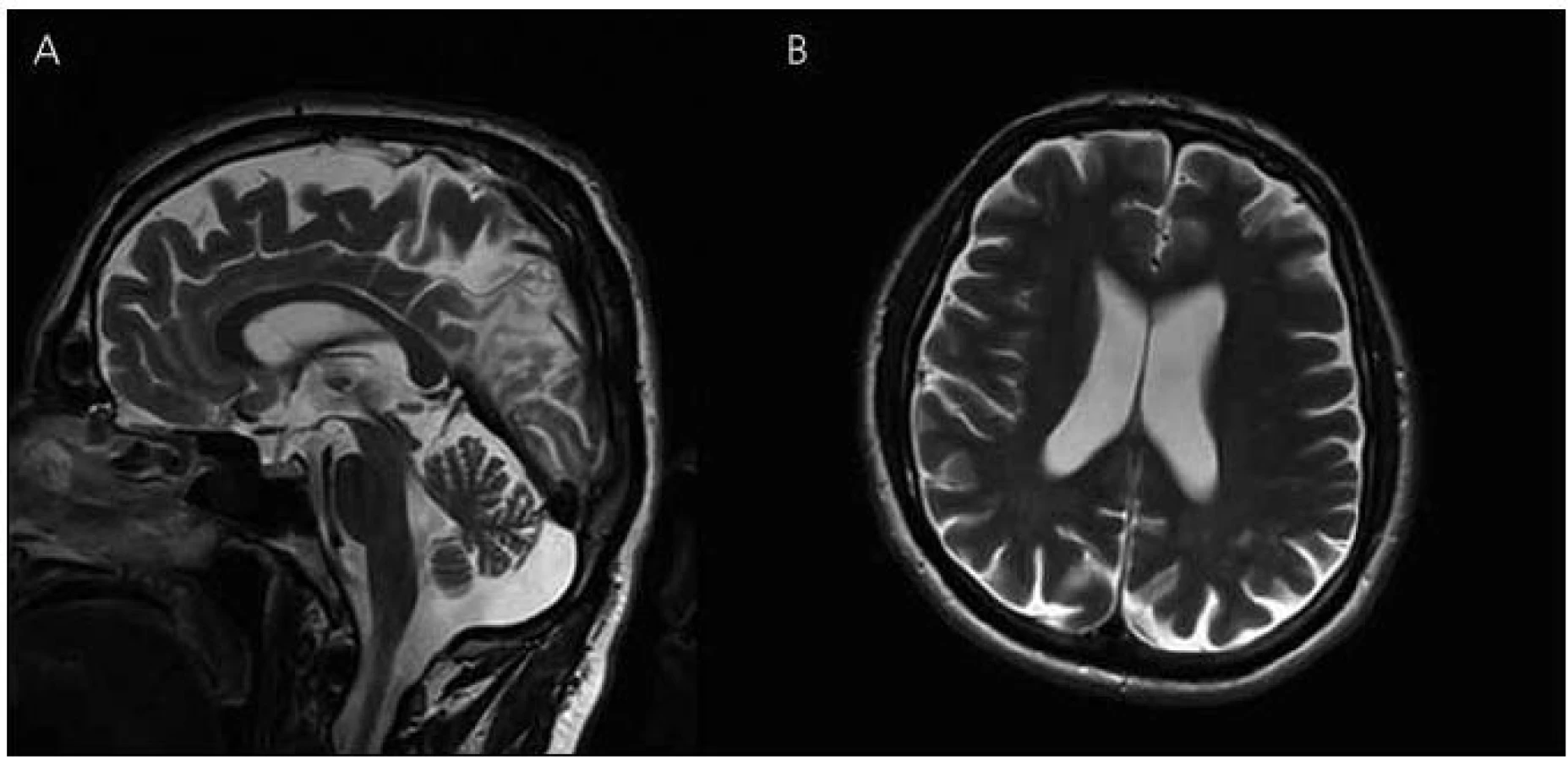
V roce 2011 se podařilo zajistit dokumentaci od dalšího žijícího stejně postiženého bratrance pacienta (obr. 1, IV-4). U pacienta začínaly první změny v chování v roce 2006 ve věku 45 let, kdy se toulal, ztrácel se při cestě domů, selhával v zaměstnání apod. Psychologické vyšetření ve 47 letech ukázalo poruchu orientace v čase, poruchu počtů, výbavnosti. V MMSE vyšetření dosáhl 20 bodů. Při vyšetření likvoru byl zjištěn normální nález, vyšetření specifických proteinů (tau protein, fosforylovaný tau protein a β amyloid) nebylo provedeno. MR mozku v roce 2008 ukazuje širší subarachnoideální prostory, difuzní atrofii mozku, ložiska gliózy v bílé hmotě obou mozkových hemisfér nespecifického charakteru (obr. 6). Byla nasazena léčba rivastigminem. Postupně i u tohoto pacienta dochází k progresi poruchy kognitivních funkcí. Nyní ve věku 50 let je již přítomna porucha řeči, obslužnosti, porucha koordinace a pády. Pacient zůstává v péči rodiny. Bylo u něho provedeno molekulárně genetické vyšetření exonu 5 genu PSEN1 metodou přímé sekvenace na genetickém analyzátoru ABI3130. Byla nalezena patogenní mutace c.415A>G (p.M139V).
Diskuze
Mutace v genu pro presenilin 1 (PSEN1) patří mezi nejčastější genetickou příčinu familiární časné formy Alzheimerovy choroby [3]. Dosud bylo popsáno v zahraniční literatuře 178 mutací v genu PSEN1 u 370 rodin s úplnou penetrancí a nástupem onemocnění mezi 25 až 65 lety (viz AD&FTD databáze mutací, www.molgen.ua.ac.be/admutations). Co se týká české populace, byla publikována jediná studie, v níž Mazura et al pátrali po mutacích v kodonu 717 exonu 17 genu APP v post mortem získaných mozkových tkáních u pacientů, u kterých se demence manifestovala před 60. rokem věku. Mutaci nalezli u dvou pacientů z 18 [14].
U probanda nalezená mutace p.M139V v genu PSEN1 byla popsána v zahraniční literatuře již u mnoha pacientů s časnou formou Alzheimerovy choroby, lze ji proto jednoznačně považovat za patogenní mutaci s autozomálně dominantním způsobem dědičnosti [13,15–17]. Nález mutace u obou bratranců je důkazem, že jejich oba otcové, kteří zemřeli v důsledku časné formy Alzheimerovy demence, nesli také tuto mutaci, stejně jako další postižení rodinní příslušníci. Z výsledků sekčního nálezu otce pacienta, jenž byl proveden v roce 1979, se dochoval pouze morfologický a histologický popis se závěrem „Alzheimerova choroba“. K upřesnění histologického nálezu na základě současných poznatků se nepodařilo získat materiál. Naše molekulárně genetické nálezy dokazují, že se u všech členů rodiny jedná o geneticky shodnou formu onemocnění, a můžeme tedy jednoznačně vyloučit možné úvahy o jiné formě demence, např. z okruhu frontotemporálních demencí, ke kterým by mohl svádět nález atrofie mozku zejména ve frontálních lalocích [18].
Mezi pacienty s různými mutacemi v genu PSEN1 existují rozdíly v nástupu příznaků onemocnění. Např. mutace p.M146V mívá nástup příznaků kolem 40 let; mutace p.M233T před 35. rokem života [16]. V literatuře najdeme rovněž publikace o de novo vzniku mutací v genu PSEN1 u sporadických časných forem Alzheimerovy choroby – např. mutace p.S170F před 30. rokem života [19]. U pacientů s mutací p.M139V jsou v závěrečných fázích onemocnění popisovány poruchy chůze, myoklonické křeče a epileptiformní stavy. Nicméně se obecně fenotyp této časné formy AD kromě věku nástupu příznaků neodlišuje od sporadické pozdní formy [2,19–21].
Náš pacient začal trpět obtížemi ve 37 letech, přičemž skutečnost, že absolvoval pouze základní vzdělání, lze považovat za rizikový faktor demence. U jeho bratrance se onemocnění manifestovalo náhle ve 45 letech. U ostatních postižených členů rodiny neznáme přesný věk nástupu onemocnění, avšak z věku úmrtí lze předpokládat jistou variabilitu věku manifestace onemocnění. Věk manifestace demence u pacientů s mutací p.M139V v literatuře kolísá mezi 32 a 48 lety [13,15–17]. Jak je z naší rodiny patrné, existuje variabilita věku nástupu onemocnění také u pacientů se stejnou mutací v rámci jedné rodiny. Jedná se o jev, který je pozorován u genetických onemocnění obecně a je způsoben tím, že v rodině se sice dědí tatáž mutace, ale jednotliví nosiči mutace se liší v jiných, tzv. modifikujících genech, jež pak mohou ovlivnit věk nástupu příznaků a průběh onemocnění. Bez vlivu jistě není ani zevní prostředí, které mají jednotliví členové odlišné. Co se týká modifikujících genů, u našeho pacienta se nabízí otázka, zda skutečnost, že je nosičem alely APOE*E4, může mít vliv na průběh onemocnění. Tuto alelu pacient zdědil od své zdravé matky. Literární údaje nasvědčují, že genotyp APOE*E4 není modifikujícím faktorem u hereditární formy Alzheimerovy choroby [21], a proto je nejpravděpodobnější, že nález alely APOE*E4 u pacienta lze považovat za náhodný, bez souvislosti s onemocněním.
Zastoupení alely APOE*E4 v kavkazské populaci je přibližně 13–17 % (přičemž frekvence homozygotů se pohybuje kolem 1 %), zatímco frekvence alely APOE*E4 u pacientů s pozdní formou Alzheimerovy choroby dosahuje 32 až 42 % i více [22,23]. Pozdní forma onemocnění (definována jako věk nástupu nad 65. roku věku) má komplexní genetický základ, kdy kromě alely APOE*E4 se zde uplatňují další nezávisle působící geny s neúplnou penetrancí, mnoho interagujících genů a také faktory zevního prostředí [24]. Nosičství alely APOE*E4 je rizikový faktor jak pro sporadickou, tak pro familiární pozdní formu onemocnění. Nosičství jedné alely APOE*E4 zvyšuje celoživotní riziko Alzheimerovy choroby přibližně trojnásobně a může způsobovat nástup onemocnění o 5–10 let dříve než u osob bez alely APOE*E4. Osoby homozygotní pro tuto alelu (APOE*E4/4) mají až 10–20 krát vyšší riziko vzniku demence oproti obecné populaci a onemocnění u nich nastupuje o 10–20 let dříve oproti osobám bez alely APOE*E4. Avšak ne všichni nosiči alely APOE*E4 onemocní, a to i v případě homozygotního stavu [23,25]. Právě proto je využití testování zdravých osob v riziku na nosičství alely APOE*E4 sporné, neboť pozitivní výsledek je spojen s psychickým stresem u testované osoby a výpovědní hodnota o riziku je nízká. Je proto obecně doporučováno testování zdravých osob na nosičství alely APOE*E4 v klinické praxi neprovádět [25].
Vyšetření genotypu APOE u zdravé matky našeho pacienta bylo provedeno po náležitém poučení s cílem zjistit, od kterého rodiče zdědil alelu APOE*E4 a zda její nosičství segreguje v rodině s onemocněním.
Znalost příčiny onemocnění v rodině nabízí možnost vyšetření zdravých osob v riziku, zda nesou vlohu k onemocnění či nikoli, tedy tzv. prediktivní či presymptomatické testování. Toto je v případě neurodegenerativních onemocnění z etického hlediska velmi složitá otázka, neboť zdravá osoba se může dozvědět, že vlohu nese, a tedy že v budoucnosti s velmi vysokou pravděpodobností onemocní, avšak tomu nelze předejít, neboť v současnosti neznáme účinnou prevenci demence. To bývá spojeno se značnou psychickou zátěží. V České republice prozatím nebylo provedeno testování zdravých osob v riziku onemocnění na nosičství mutace v genu PSEN1 (v dané rodině o toto příbuzní neprojevili zájem), avšak bylo by vhodné se v případě zájmu řídit pravidly, která platí pro Huntingtonovu choreu [26]. Zde je možné zdravé osoby prediktivně testovat až poté, co prošly mezinárodně unifikovaným protokolem vyšetření, který zahrnuje opakovaná vyšetření neurologická, psychologická, psychiatrická a genetická [27]. Vyšetření mutace je možné provést až poté, co jsou psychologem a psychiatrem uznáni za způsobilé k prediktivnímu testování na nosičství vlohy k neurodegenerativnímu onemocnění.
U našeho případu bylo důležité, že se matka pacienta, která tušila možný rozvoj demence u svého syna vzhledem k rodinné anamnéze, sama dožadovala časného neurologického vyšetření syna a upozorňovala na možnou rodinnou zátěž. Před neurologickým vyšetřením byl pacient už opakovaně vyšetřován pro projevy kognitivního deficitu v psychiatrické ambulanci. Přesto byla léčba inhibitory acetylcholinesterázy nasazena až při projevech výrazného kognitivního postižení. Nicméně lze předpokládat, že léčba pacienta inhibitory acetylcholinesterázy zpomalila další progresi demence a umožnila lepší úroveň aktivit denního života pacienta.
V objektivním neurologickém vyšetření v roce 2003 byly u pacienta popsány prvky mimovolních pohybů typu chorey, zvláště na horních končetinách. Bylo proto indikováno vyšetření Huntingtonovy chorey, s negativním výsledkem. Tyto projevy určitě nelze považovat za abnormální manifestaci demence v souvislosti s mutací v genu PSEN1. Těmito pohyby pacient na počátku choroby maskoval anxietu a úzkost, která se projevovala hlavně v sociálních kontaktech.
Zobrazovací metody, jako je počítačová tomografie (CT) nebo lépe MR, pomáhají v diferenciální diagnostice demencí k vyloučení strukturálních lézí mozku typu expanzivních procesů a cévních změn vedoucích k syndromu demence. Tato vyšetření mohou prokázat atrofii mozku především v oblasti temporální a rozšíření mozkových komor, které jsou výraznější než u fyziologického procesu stárnutí. Mohou zde být i známky mírné angiopatie v bílé hmotě hemisfér a v podkoří, protože amyloid se může ukládat i do cévní stěny [28]. Také v námi popisovaném případě došlo během osmiletého průběhu onemocnění k výrazné atrofii mozku a rozšíření mozkových komor, které provázelo deterioraci kognitivních funkcí. Uvedené demyelinizační změny v bílé hmoty jsou nespecifického charakteru. Naše zobrazení MR má své limitace, protože první vyšetření bylo provedeno ještě na přístroji MR, kde nebylo možné některé sekvence včetně volumometrie hippokampu provést. U druhého vyšetření mají koronární řezy pro neklid pacienta pohybové artefakty, proto nebyly použity. Vyšetření muselo být předčasně ukončeno pro neklid pacienta. U druhého vyšetřovaného pacienta v rodině byly nalezeny na MR mozku také atrofické změny mozku.
Závěr
Hereditární predispozice k časné formě Alzheimerovy demence způsobená zárodečnými mutacemi v genu PSEN1 je vzácné genetické onemocnění přenášené autozomálně dominantně v postižených rodinách. Genetické vyšetření při výskytu časné formy demence v rodině je možné také v České republice (v případě zájmu o vyšetření je možné se obrátit na Laboratoř DNA diagnostiky Oddělení lékařské genetiky ve Fakultní nemocnici Ostrava). V současné době je jeho význam především diagnostický. Pravděpodobně však až v budoucnosti bude mít toto testování větší význam vzhledem k momentálně omezeným možnostem preventivní léčby v postižených rodinách.
MUDr.
Petra Bártová, Ph.D.
Neurologická
klinika
OU
a FN Ostrava
tř.
17. listopadu 1790
708
52 Ostrava
e-mail:
petrabartova@seznam.cz
Přijato
k recenzi: 30. 11. 2010
Přijato
do tisku: 8. 3. 2011
Zdroje
1. Ferri CP, Prince M, Brayne C, Brodaty H, Fratiglioni L, Ganguli M et al. Global prevalence of dementia: a Delphi consensus study. Lancet 2005; 366(9503): 2112–2117.
2. Jirák R, Koukolík F. Demence. Praha: Galén 2002: 106–144.
3. Bertram L, Lill CM, Tanzi RE. The genetics of Alzheimer disease: back to the future. Neuron 2010; 68(2): 270–281.
4. Bekris LM, Yu CE, Bird TD, Tsuang DW. Genetics of Alzheimer disease. J Geriatr Psychiatry Neurol 2010; 23(4): 213–227.
5. Kowalska A. Genetics of dementias. Part 4: a spectrum of mutations responsible for the familial autosomal dominant form of Alzheimer’s disease. Postepy Hig Med Dosw 2009; 63: 583–591.
6. Vetrivel KS, Zhang YW, Xu H, Thinakaran G. Pathological and physiological functions of presenilins. Mol Neurodegener 2006; 1: 4.
7. Vetrivel KS, Thinakaran G. Membrane rafts in Alzheimer’s disease beta-amyloid production. Biochim Biophys Acta 2010; 1801(8): 860–867.
8. Jozwiak K, Zekanowski C, Filipek S. Linear patterns of Alzheimer’s disease mutations along α-helices of presenilins as a tool for PS-1 model construction. J Neurochem 2006; 98(5): 1560–1572.
9. Das HK. Transcriptional regulation of the presenilin-1 gene: implication in Alzheimer’s disease. Front Biosci 2008; 13: 822–832.
10. Folstein MF, Folstein SE, McHugh PR. Mini-mental state: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975; 12(3): 189–198.
11. Dubois B, Feldman HH, Jacova C, Dekosky ST, Barberger-Gateau P, Cummings J et al. Research criteria for the diagnosis of Alzheimer’s disease: revising the NINCDS-ADRDA criteria. Lancet Neurol 2007; 6(8): 734–746.
12. Ressner P, Hort J, Rektorová I, Bartoš A, Rusina R, Línek V et al. Doporučené postupy pro diagnostiku Alzheimerovy nemoci a dalších nemocí spojených s demencí. Cesk Slov Neurol N 2008; 71/104(4): 494–501.
13. Finckh U, Müller-Thomsen T, Mann U, Eggers C, Marksteiner J, Meins W et al. High prevalence of pathogenic mutations in patients with early-onset dementia detected by sequence analyses of four different genes. Am J Hum Genet 2000; 66(1): 110–117.
14. Mazura I, Koukolík F, Jirák R. Molecular genetic analysis of Alzheimer’s dementia in the Czech population. The APP-717 mutation in the gene for amyloid protein precursor. Cas Lek Cesk 1999; 138(3): 75–77.
15. Hüll M, Fiebich BL, Dykierek P, Schmidtke K, Nitzsche E, Orszagh M et al. Early-onset Alzheimer’s disease due to mutations of the presenilin-1 gene on chromosome 14: a 7-year follow-up of a patient with a mutation at codon 139. Eur Arch Psychiatry Clin Neurosci 1998; 248(3): 123–129.
16. Zekanowski C, Styczyńska M, Pepłońska B, Gabryelewicz T, Religa D, Ilkowski J et al. Mutations in presenilin 1, presenilin 2 and amyloid precursor protein genes in patients with early-onset Alzheimer’s disease in Poland. Exp Neurol 2003; 184(2): 991–996.
17. Fox NC, Kennedy AM, Harvey RJ, Lantos PL, Roques PK, Collinge J et al. Clinicopathological features of familial Alzheimer’s disease associated with the M139V mutation in the presenilin 1 gene. Pedigree but not mutation specific age at onset provides evidence for a further genetic factor. Brain 1997; 120(Pt 3): 491–501.
18. Rektorová I. Frontotemporální lobární degenerace – diagnosa z neuro-psychiatrického pomezí. Neurol pro praxi 2006; 4: 199–202.
19. Golan MP, Styczyńska M, Jóźwiak K, Walecki J, Maruszak A, Pniewski J et al. Early-onset Alzheimer’s disease with a de novo mutation in the presenilin 1 gene. Exp Neurol 2007; 208(2): 264–268.
20. Koukolík F, Jirák R. Alzheierova nemoc a další demence. Praha: Grada 1998: 89–90.
21. Hort J, O’Brien JT, Gainotti G, Pirttila T, Popescu BO, Rektorová I et al. EFNS Scientist Panel on Dementia. EFNS guidelines for the diagnosis and management of Alzheimer’s disease. Eur J Neurol 2010; 17(10): 1236–1248.
22. Styczyńska M, Strosznajder JB, Religa D, Chodakowska-Zebrowska M, Pfeffer A, Gabryelewicz T et al. Association between genetic and environmental factors and the risk of Alzheimer’s disease. Folia Neuropathol 2008; 46(4): 249–254.
23. Hsiung GY, Sadovnick AD, Feldman H. Apolipoprotein E ε4 genotype as a risk factor for cognitive decline and dementia: data from the Canadian Study of Health and Aging. CMAJ 2004; 171(8): 863–867.
24. Nussbaum RL, McInnes RR, Willard HF. Thompson & Thompson: Genetics in Medicine. 6th ed. Philadelphia: WB Sounders Company 2001: 203–247.
25. Firth HV, Hurst JA. Oxford Desk Reference: Clinical Genetics. Oxford: Oxford University Press 2005: 296–297.
26. International Huntington Association (IHA) and the World Federation of Neurology (WFN) Research Group of Huntington’s Chorea. Guidelines for the molecular genetics predictive test in Huntington’s disease. Neurology 1994; 44(8): 1533–1536.
27. Coustasse A, Pekar A, Sikula A, Lurie S. Ethical considerations of genetic presymptomatic testing for Huntington’s disease. J Hosp Mark Public Relations 2009; 19(2): 129–141.
28. Hort J, Rusina J. Paměť a její poruchy. Praha: Maxdorf 2007: 158–177.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2011 Číslo 5
Nejčtenější v tomto čísle
- Vývojová porucha koordinace – vývojová dyspraxie
- Kognitivní evokované potenciály
- Progredující spasticita, kognitivní deficit a nevýbavné kortikální motorické evokované potenciály jako klinické příznaky pravděpodobné primární laterální sklerózy – kazuistika
- Zkušenosti s evakuací chronického subdurálního hematomu z návrtu kalvy