
Maligní tumor z pochvy periferního nervu – dvě kazuistiky
Malignant Peripheral Nerve She ath Tumor – Two Case Reports
Malignant peripheral nerve she ath tumor (MPNST) is a highly malignant tumor of the peripheral nervo us system, which is likely to reccur locally or metastatize. Its incidence is higher in pati ents with ne urofibromatosis type 1 (NF‑1). We present two case reports of pati ents with this rare tumor. First pati ent, 74- ye ar old woman, underwent ro utine chest exam which disclosed an expansive lesi on of the left chest cavity. Magnetic resonance imaging appe arance suggested a Th3 schwannoma. However, histopathological exam after parti al resecti on disclosed MPNST with proliferative potenti al of 25 %. The pati ent di ed six months later due to tumor progressi on. Second pati ent, 35- ye ar old woman with NF‑1 noticed enlargement of a known lump of the right calf. Histopathological exam after parti al resecti on disclosed ne urofibroma with signs of MPNST. Control magnetic resonance imaging reve aled massive tumor progressi on; the pati ent underwent limb amputati on. The pati ent has been in remissi on during 20 months follow‑up. Apart from these case reports the paper revi ews the literature as well as clinical and therape utical aspects of this tumor.
Key words:
malignant peripheral nerve she ath tumor – malignant schwannoma – ne urofibromatosis – soft tissue sarcoma
Autoři:
V. Beneš lll 1; F. Kramář 2; P. Hrabal 3; M. Kaiser 1; P. Buchvald 1
Působiště autorů:
Neurochirurgické oddělení, Krajská nemocnice Liberec, 2Neurochirurgická klinika 1. LF UK, IPVZ a ÚVN Praha, 3Oddělení patologie ÚVN Praha
1
Vyšlo v časopise:
Cesk Slov Neurol N 2009; 72/105(2): 163-167
Kategorie:
Kazuistika
Souhrn
Maligní tumor z pochvy periferního nervu (MPNST) představuje vysoce maligní nádor periferního nervového systému, který mnohdy recidivuje a zakládá vzdálené metastázy. Častější výskyt byl zaznamenán u paci entů s ne urofibromatózo u typu 1. Předloženy jso u dvě kazuistiky paci entek s tímto vzácným tumorem. U první paci entky, 74leté ženy rutinní vyšetření hrudníku odhalilo expanzi v levém plicním poli. Z následné magnetické rezonance bylo usuzováno na schwannom Th3, nicméně histopatologické vyšetření po parci ální resekci prokázalo MPNST s proliferační aktivito u 25 %. Paci entka zemřela za šest měsíců na generalizaci nádorového procesu. Druhá paci entka, 35letá žena s ne urofibromatózo u typu 1, si povšimla nárůstu již známé rezistence na pravém lýtku. Histopatologický rozbor po parci ální resekci však prokázal ne urofibrom místy se znaky MPNST. Kontrolní magnetická rezonance ukázala objemno u recidivu; následovala amputace končetiny. Paci entka je 20 měsíců od prvního výkonu v remisi. Vedle těchto kazuistik práce přináší přehled literatury a diskutuje klinické a terape utické aspekty tohoto nádoru.
Klíčová slova:
maligní tumor periferního nervového systému – maligní schwannom – ne urofibromatóza – sarkom měkkých tkání
Úvod
Maligní tumor z pochvy periferního nervu představuje vzácný nádor periferního nervového systému. Dle World Health Organization se jedná o maligní tumor vycházející z této části periferního nervu nebo se diferencující jejím směrem. Vyjmuty z této definice jsou tumory vyrůstající z epineuria či cévního zásobení periferního nervu [1]. Dříve používané termíny jako maligní schwannom, neurofibrosarkom, neurogenní sarkom či maligní neurilemmom jsou dnes opouštěny ve prospěch anglického termínu Malignant Peripheral Nerve Sheath Tumor (MPNST).
MPNST se nejčastěji nalézá v oblasti trupu (50 %), končetin (30 %) nebo hlavy a krku (20 %) [2,3], v naší literatuře byl popsán i intrakraniální MPNST [4]. Baehring et al doporučují rozlišovat lokalizaci proximální (míšní kořen, plexus) a lokalizaci distální (periferní nerv, terminální vlákna), lépe totiž odráží možnost radikální resekce [5]. Její dosažení je prakticky nemožné v případě paraspinální lokalizace tumoru [6]. Předkládáme kazuistiku pacientky s MPNST právě v této lokalizaci. Dále pak vzhledem k častějšímu výskytu MPNST u pacientů s neurofibromatózou typu 1 (NF-1) na příkladě pacientky s tímto genetickým syndromem demonstrujeme agresivní chování MPNST v této populaci pacientů. Vedle těchto kazuistik přinášíme přehled literatury a diskutujeme o klinických a terapeutických aspektech tohoto vzácného tumoru.
Kazuistika 1
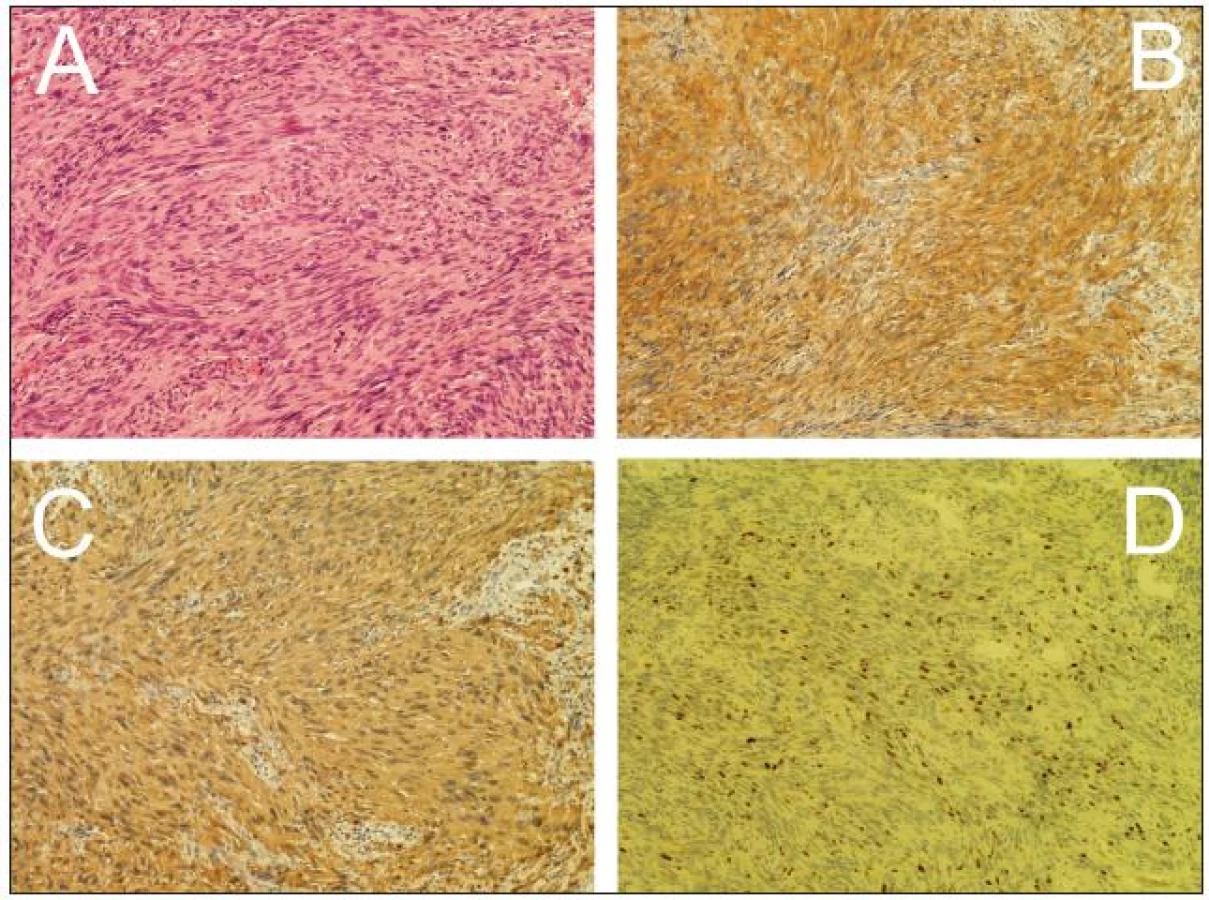
Žena, 74 let, podstoupila z interní indikace rentgenové vyšetření hrudníku. To odhalilo expanzi levého plicního pole. Z doplněné magnetické rezonance (MR), která prokázala nehomogenně se sytící sutkovitou expanzi Th3 vlevo, bylo usuzováno na schwannom (obr. 1). Pacientka podstoupila parciální resekci, nádor se makroskopicky jevil jako schwannom. Nicméně histologicky se jednalo o značně buněčný nádor z vřetenitých buněk s protáhlými jádry uspořádaných do fasciklů exprimující S-100 protein a neuron-specifickou enolázu (NSE). Přítomnost četných mitóz a MIB-1 proliferační index kolem 25 % svědčily pro vysokou proliferační aktivitu nádoru (obr. 2). Na základě těchto nálezů byl nádor klasifikován jako MPNST. Následně pacientka podstoupila aktinoterapii dané oblasti. Zemřela necelých šest měsíců po operaci na generalizaci nádorového onemocnění.
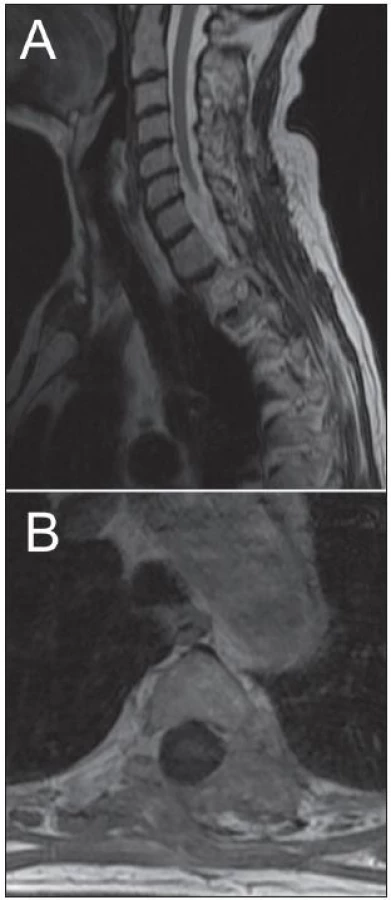
Kazuistika 2
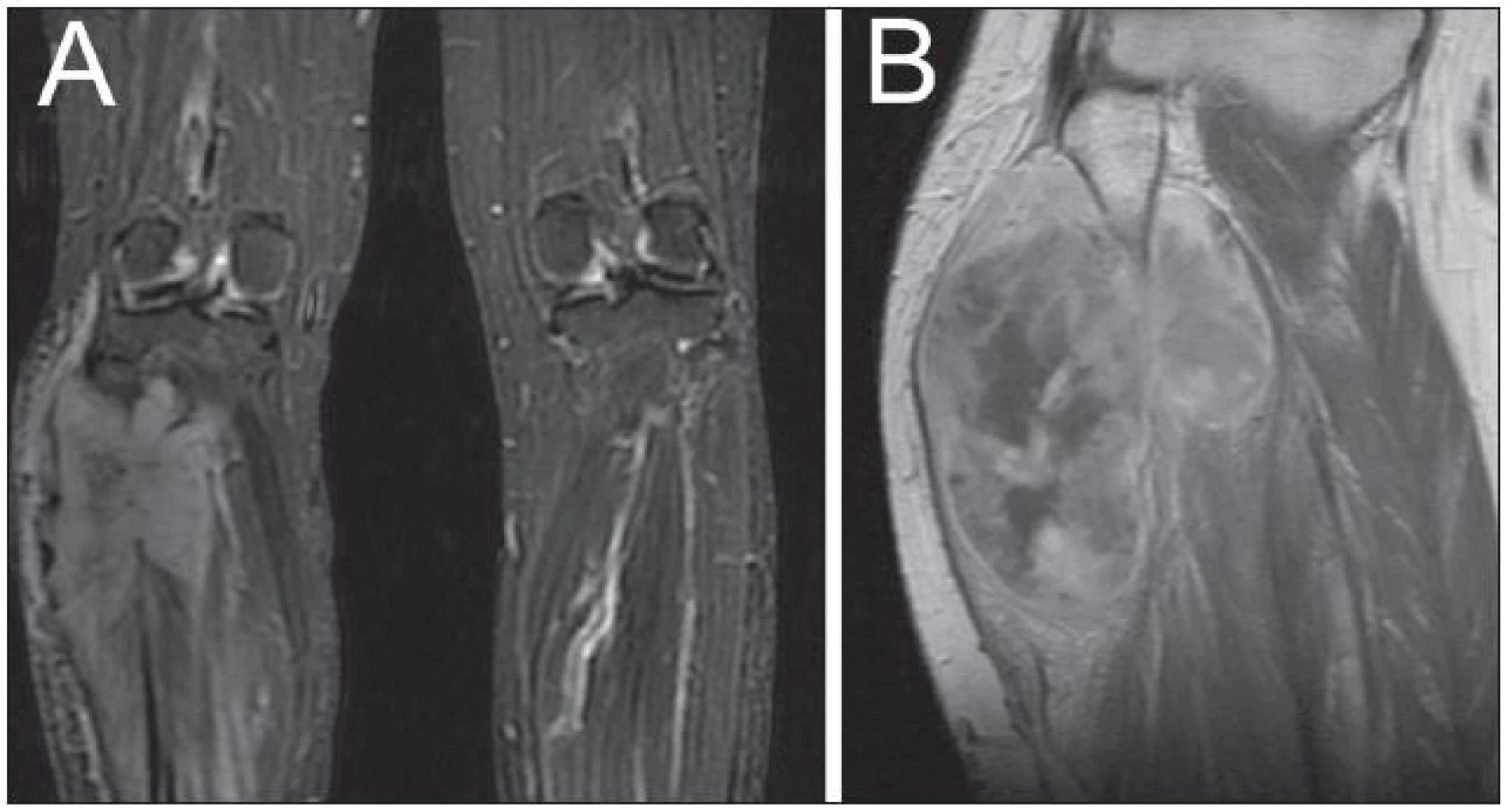
Žena, 35 let, postižená NF-1, si povšimla postupného nárůstu již dříve známé rezistence na pravém lýtku a rovněž rozvoje hypestezie na plosce pravé dolní končetiny. MR dolních končetin prokázalo expanzi v oblasti lýtka vpravo (obr. 3A), vzhledem k základnímu onemocnění bylo pomýšleno na neurofibrom. Podstoupila parciální resekci tumoru, který se v histologickém obraze jevil jako celulární schwannom, ale místy se svými rysy blížil diagnóze MPNST. Po operaci bylo rozhodnuto o prozatímní observaci. Na kontrolním MR dolních končetin za tři měsíce byla patrná objemná recidiva procesu (obr. 3B). Následovala další subtotální resekce. Jednalo se o vysoce buněčný tumor fascikulárního uspořádání s ložiskově naznačeným myxoidním charakterem. Imunohistochemická pozitivita na S-100 protein spolu s četnými mitózami a MIB-1 proliferačním indexem kolem 25 % potvrdily diagnózu MPNST (obr. 4). Pacientka posléze podstoupila na doporučení onkologa vysokou amputaci pravé dolní končetiny. V současné době, 20 měsíců od první operace, je pacientka bez zjistitelných známek recidivy nádorového procesu.

Diskuze
MPNST, jehož incidence se v běžné populaci odhaduje na 0,001 % [3,5], postihuje muže zhruba stejně často jako ženy, obvykle ve čtvrtém až šestém deceniu [3,7,8]. Incidence je vyšší u pacientů s NF-1 (4–5 %) [3,5,9]. NF-1 je nejčastější genetický syndrom podmiňující vznik nádorů. NF-1 se vyskytuje s frekvencí 1 : 3 000 nově narozených [10]. Gen NF-1 na 17. chromozomu kóduje protein neurofibromin; při jeho inaktivaci dochází k nekontrolované proliferaci buněk zapříčiněné vysokou aktivitou v systému Ras-GTP [11]. NF-1 funguje jako klasický tumor supresorový gen. Pacienti s NF-1 mají od narození jednu defektní kopii a pokud následná mutace vyřadí druhou, dojde ke splnění podmínek tumorigeneze podle známého Knudsonova modelu [12]. U pacientů s NF‑1 toto vede ke vzniku četných benigních neurofibromů. Za jejich maligní transformaci velmi pravděpodobně nesou odpovědnost následné mutace v dalších genech, např. p53, RB1 či mdm2 [13–18]. Při nepřítomnosti zděděné nefunkční kopie NF‑1 genu je popsaná sekvence velmi nepravděpodobná, což vysvětluje až 4 000× nižší incidenci MPNST v běžné populaci. MPNST se u pacientů s NF‑1 vyskytují zhruba o 10 let dříve, bývají větší, proximálně lokalizované, multifokální a jejich prognóza je závažnější [2,3,8,19]. Časná a objemná recidiva tumoru u naší pacientky s NF‑1 dobře ilustruje tento prognosticky nepříznivý trend. Podobně jako v jejím případě může na maligní transformaci upozornit růst stávajícího neurofibromu nebo nově vzniklá bolest či neurologický deficit [3,8,20,21], přínosná je v tomto případě pozitronová emisní tomografie (PET); vyšší metabolický obrat glukózy svědčí pro maligní lézi [20,22]. Celoživotní riziko vzniku MPNST se u pacientů s NF‑1 uvádí jako 10% [22]. Přibližně u desetiny všech MPNST lze nalézt souvislost s předchozí radioterapií dané oblasti, u pacientů s NF‑1 by se tudíž radioterapie měla používat velmi obezřetně [22–24]. Dlouhodobé sledování po radioterapii je naprostá nezbytnost, byly popsány MPNST vzniklé i za více než 25 let [23].
Vedle již zmíněné symptomatologie u NF‑1 na sebe MPNST nejčastěji upozorní jako narůstající zduření lokalizované v průběhu nervu. V případě centrální lokalizace se poté jedná o symptomatologii vyplývající z útlaku míchy nebo míšních kořenů. Avšak byly popsány i MPNST náhodně odhalené [5], stejně jako v případě naší první pacientky. Vedle pečlivého neurologického vyšetření je zapotřebí věnovat pozornost známým projevům neurofibromatózy (skvrny café au lait, kožní neurofibromy, hyperpigmentace, Lischovy uzlíky atd.) [21,25]. Diagnostický algoritmus by měl zahrnovat ultrazvuk, při podezření na kostní invazi výpočetní tomografii (CT), a samozřejmě MR, která dokonale ozřejmí lokální anatomické poměry. Vzhledem k častému metastatickému postižení (až čtvrtina pacientů v době diagnózy [8]) je vhodné doplnit prostý snímek hrudníku a sonografii břicha, při pozitivním nálezu poté CT nebo PET CT. MR může přispět pro odlišení benigního od maligně transformovaného neurofibromu přítomností tzv. target sign. Tento by měl být přítomen jako hlavní součást tumoru. Jedná se o centrální hypointenzní oblast obklopenou hyperintenzním lemem na T2 vážených sekvencích. Histologicky centrální oblasti odpovídá nahromadění Schwannových buněk a kolagenu, okolí poté tvoří myxoidní stroma. Jeho nepřítomnost nebo pouze okrajový výskyt poté hovoří pro MPNST [26]. U obou pacientek jsme se předoperačně přikláněli ke klasickému schwannomu, z kontrolního MR druhé pacientky je však jasně patrné agresivní chování tumoru.
Konečnou diagnózu s určitostí stanoví až vyšetření histopatologické. MPNST se jeví jako vysoce celulární tumor uspořádaný do fasciklů. Připomíná svou stavbou sarkomy měkkých tkání, nejčastěji pak fibrosarkom [27]. Buňky obvykle vřetenovitého tvaru mívají neostré hranice a jádra s jemným tečkovitým chromatinem [28,29]. Divergentní diferenciace s příměsí epiteliálních, glandulárních, chondrosarkomatózních, rabdomyosarkomatózních či osteosarkomatózních elementů není výjimečná [3,7,30,31]. Imunohistochemicky může MPNST vykazovat pozitivitu pro S-100, NSE, vimentin či Leu 7 [2,30]. Negativita těchto imunomarkerů jeho diagnózu nevylučuje, naopak hovoří pro nízký stupeň diferenciace, a tím pádem i horší prognózu [6]. Výše MIB-1 proliferačního indexu se pohybuje mezi 10–30 %, p53 poté mezi 25–80 % [2]. Kritéria malignity dále zahrnují invazi do okolních tkání, cév, nukleární pleomorfizmus, nekrózu a přítomnost mitotických figur. Hruban et al, stejně jako Wong et al nalezli signifikantní asociaci mezi prognózou a počtem mitóz [7,19] nebo přítomností nekróz [19]. Naopak Kourea et al u těchto dvou parametrů signifikantní asociaci nenalezli [27]. Řada autorů používá různá rozdělení na jednotlivé „grade“ [8,19,27,32], avšak dosud neexistuje obecně přijímaná histologická klasifikace, která by dokázala spolehlivě předpovědět prognózu [33]. Histopatologické vyšetření obou prezentovaných pacientek splňovalo výše uvedená kritéria, vysoká proliferační aktivita byla následně potvrzena generalizací, respektive časnou a objemnou recidivou.
Podobně jako u sarkomů měkkých tkání je základním pilířem léčby radikální bloková resekce tumoru s histologicky negativními okraji [2,3,5,7,8,19,24,31,34–37], a to i za cenu amputace končetiny [31], ke které jsme neváhali přistoupit. Při pozitivním nálezu v okrajích resekce se významně snižuje pětileté přežití, a to z 82 na 41 % [8] či pouze na 22 % [19]. Resekabilita odvisí od lokalizace a pohybuje se od 20 % v případě paraspinálních MPNST do 95 % u končetinových MPNST [3,7,19,27,31,32]. Mezi další nepříznivé faktory se mimo jiné počítá velikost tumoru nad 5cm [3,5,8,34], asociace s NF‑1 [3,7,8,24,31], předchozí radioterapie dané oblasti [19,23,38], neradikální resekce [3,5] a subdiafragmatická či nitrohrudní lokalizace [27].
Cílem resekce je dosažení lokální kontroly nemoci, recidiva významně snižuje přežití [7,8]. U částečné resekce adjuvantní radioterapie, stejně jako intersticiální brachyterapie, zlepšuje lokální kontrolu nemoci a tím i celkovou prognózu [19]. Anghileri et al doporučují radioterapii u všech pacientů [8]. Dávka se pak pohybuje mezi 45–65 Gy v závislosti na citlivosti okolních tkání [2,8,19,39]. Role chemoterapie zůstává kontroverzní [3,7,19,27,32]. Pokud nejsou přítomny metastázy a resekce je radikální, nemá větší přínos [5].
Metastatické postižení v průběhu nemoci se objevuje u 26–65 % pacientů [3,7,8,19,27,31,32], signifikantně častěji pak u pacientů s lokální recidivou a větším tumorem [8]. Nejčastějším orgánem postiženým metastázami jsou plíce následované kostmi [7,8].
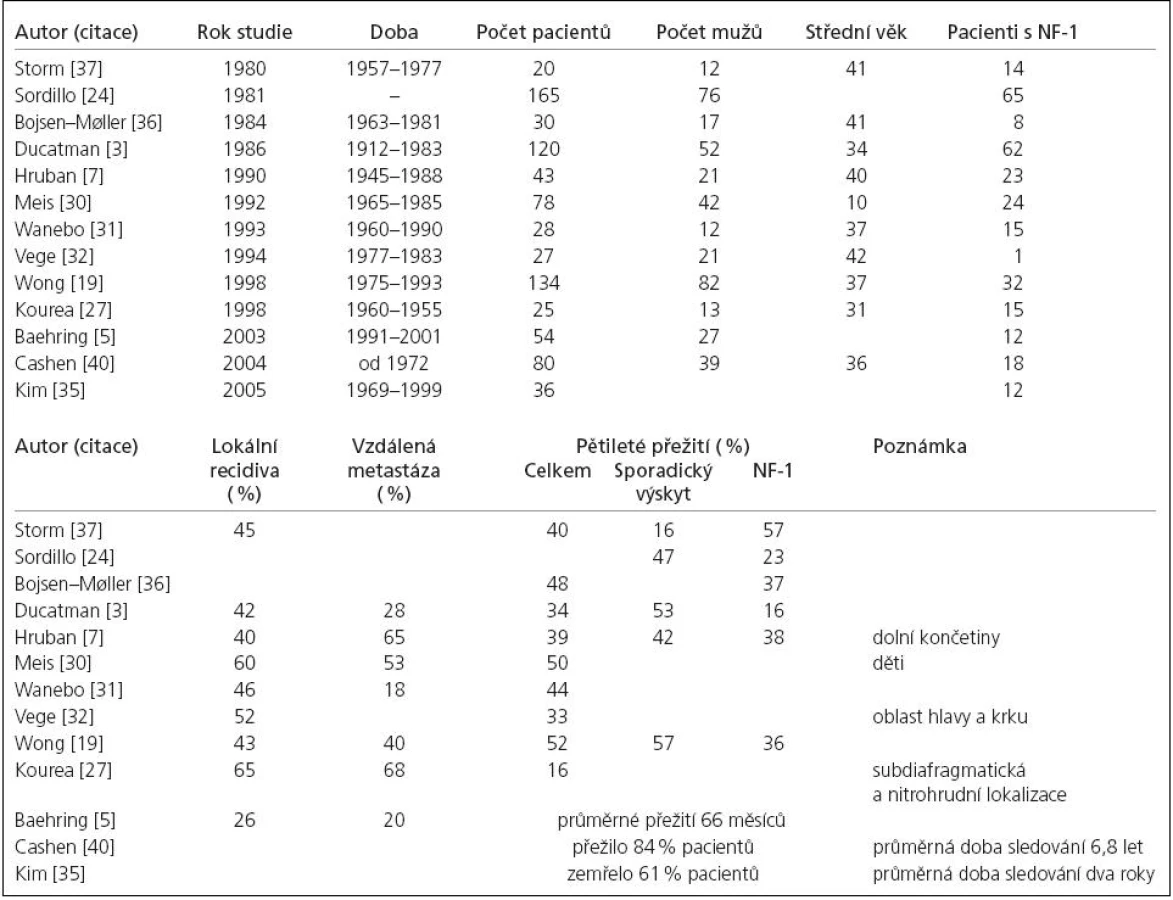
Celková prognóza pacienta postiženého MPNST je velmi závažná. Pětileté přežití se v největších studiích pohybovalo mezi 16–60 % [3,5,7,8,19,24,27,30–32,35–37]. Poněkud stranou stojí série Cashenova, 84 % pacientů přežilo při průměrném sledování 6,8 let. Autor se domnívá, že za těmito vynikajícími výsledky stojí radio- a chemoterapie, časnější diagnóza a užití moderních zobrazovacích metod. Jasný léčebný protokol bohužel neudává [40]. Přehled demografických dat, souvislosti s NF‑1, četnost lokálních recidiv, vzdálených metastáz a pětiletého přežití ve významných studiích ukazuje tab. 1.
Závěr
Diagnóza MPNST je prognosticky velmi závažná. Jedná se o vysoce maligní nádor, který často recidivuje a metastazuje. Pětileté přežití se pohybuje okolo 45 %. Zvýšenou pozornost je třeba věnovat pacientům s NF‑1, u nichž může MPNST vzniknout maligní transformací benigního neurofibromu.
MUDr. Vladimír Beneš
Neurochirurgické oddělení
Krajská nemocnice Liberec
Husova 10
460 63 Liberec
e‑mail:
vladimir.benes@nemlib.cz
Zdroje
1. Wo odruff JM, Ko ure a HP, Lo uis DN, Scheitha uer BW. Malignant peripheral nerve she ath tumo ur (MPNST). In: Kleihues P, Cavenee WK (eds). Pathology and genetics of tumo urs of the nervo us system. Lyon: IARCPress 2000: 172– 174.
2. Stark AM, Buhl R, Hugo HH, Mehdorn HM. Malignant peripheral nerve she ath tumo urs – report of 8 cases and revi ew of the literature. Acta Ne urochir (Wi en) 2001; 143(4): 357– 364.
3. Ducatman BS, Scheitha uer BW, Pi epgras DG, Reiman HM, Ilstrup DM. Malignant peripheral nerve she ath tumors. A clinicopathologic study of 120 cases. Cancer 1986; 57(10): 2006– 2021.
4. Bludovský D, Hes O, Choc M, Michal M. Intracerebrální supratentori ální maligní schwannom: Kazuistika. Cesk Slov Ne urol N 2001; 68/ 101(2): 120– 123.
5. Baehring JM, Betensky RA, Batchelor TT. Malignant peripheral nerve she ath tumor: the clinical spectrum and o utcome of tre atment. Ne urology 2003; 61(5): 696– 698.
6. Seppälä MT, Halti a MJ. Spinal malignant nerve- she ath tumor or cellular schwannoma? A striking difference in prognosis. J Ne urosurg 1993; 79(4): 528– 532.
7. Hruban RH, Shi u MH, Seni e RT, Wo odruff JM. Malignant peripheral nerve she ath tumors of the buttock and lower extremity. A study of 43 cases. Cancer 1990; 66(6): 1253– 1265.
8. Anghileri M, Miceli R, Fi ore M, Mari ani L, Ferrari A, Mussi C et al. Malignant peripheral nerve she ath tumors: prognostic factors and survival in a seri es of pati ents tre ated at a single instituti on. Cancer 2006; 107(5): 1065– 1074.
9. Tucker T, Wolkenstein P, Revuz J, Zeller J, Fri edman JM. Associ ati on between benign and malignant peripheral nerve she ath tumors in NF1. Ne urology 2005; 65(2): 205– 211.
10. Huson SM, Harper PS, Compston DA. Von Recklingha usen ne urofibromatosis. A clinical and populati on study in so uth- e ast Wales. Brain 1988; 111(6): 1355– 1381.
11. Schwarz J, Belzberg AJ. Malignant peripheral nerve she ath tumors in the setting of segmental ne urofibromatosis. Case report. J Ne urosurg 2000; 92(2): 342– 346.
12. Knudson AG jr. Mutati on and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA 1971; 68(4): 820– 823.
13. Kawai A, Noguchi M, Beppu Y, Yokoyama R, Mukai K, Hirohashi S et al. Nucle ar immunore acti on of p53 protein in soft tissue sarcomas. A possible prognostic factor. Cancer 1994; 73(10): 2499– 2505.
14. Cance WG, Brennan MF, Dudas ME, Hu ang CM, Cordon- Cardo C. Altered expressi on of the retinoblastoma gene product in human sarcomas. N Engl J Med 1990; 323(21): 1457– 1462.
15. Cordon- Cardo C, Latres E, Drobnjak M, Oliva MR, Pollack D, Wo odruff JM et al. Molecular abnormaliti es of mdm2 and p53 genes in adult soft tissue sarcomas. Cancer Res 1994; 54(3): 794– 799.
16. Halling KC, Scheitha uer BW, Halling AC, Nascimento AG, Zi esmer SC, Roche PC et al. p53 expressi on in ne urofibroma and malignant peripheral nerve she ath tumor. An immunohistochemical study of sporadic and NF1‑associ ated tumors. Am J Clin Pathol 1996; 106(3): 282– 288.
17. Kindblom LG, Ahldén M, Meis- Kindblom JM, Stenman G. Immunohistochemical and molecular analysis of p53, MDM2, proliferating cell nucle ar antigen and Ki67 in benign and malignant peripheral nerve she ath tumo urs. Virchows Arch 1995; 427(1): 19– 26.
18. Legi us E, Di erick H, Wu R, Hall BK, Marynen P, Cassiman JJ et al. TP53 mutati ons are frequent in malignant NF1 tumors. Genes Chromosomes Cancer 1994; 10(4): 250– 255.
19. Wong WW, Hirose T, Scheitha uer BW, Schild SE, Gunderson LL. Malignant peripheral nerve she ath tumor: Analysis of tre atment o utcome. Int J Radi ati on Oncology Bi ol Phys 1998; 42(2): 351– 360.
20. Ferner RE, Lucas JD, O’Doherty MJ, Hughes RAC, Smith MA, Cronin BF et al. Evalu ati on of (18)fluorode oxyglucose positron emissi on tomography ((18)FDG PET) in the detecti on of malignant peripheral nerve she ath tumo urs arising from within plexiform ne urofibromas in ne urofibromatosis 1. J Ne urol Ne urosurg Psychi atry 2000; 68(3): 353– 357.
21. Ferner RE, Gutmann DH. Internati onal consensus statement on malignant peripheral nerve she ath tumors in ne urofibromatosis. Cancer Res 2002; 62(5): 1573– 1577.
22. Evans DGR, Baser ME, McGa ughran J, Sharif S, Howard E, Moran A. Malignant peripheral nerve she ath tumo urs in ne urofibromatosis 1. J Med Genet 2002; 39(5): 311– 314.
23. Ducatman BS, Scheitha uer BW. Postirradi ati on ne urofibrosarcoma. Cancer 1983; 51(6): 1028– 1033.
24. Sordillo PP, Helson L, Hajdu SI, Magill GB, Kosloff C, Golbey RB et al. Malignant schwannoma – clinical characteristics, survival and response to therapy. Cancer 1981; 47(10): 2503– 2509.
25. Kozler P, Kramář F. Famili ární nádorové syndromy postihující nervový systém. In: Kozler P et al (eds). Intrakrani ální nádory. 1st ed. Praha: Galén, Karolinum 2007: 175– 182.
26. Bhargava R, Parham DM, Lasater OE, Chari RS, Chen G,Fletcher BD. MR imaging differenti ati on of benign and malignant peripheral nerve she ath tumors: use of the target sign. Pedi atr Radi ol 1997; 27(2): 124– 129.
27. Ko ure a HP, Bilsky MH, Le ung DH, Lewis JJ, Wo odruff JM. Subdi aphragmatic and intrathoracic paraspinal malignant peripheral nerve she ath tumors. A clinicopathological study of 25 pati ents and 26 tumors. Cancer 1998; 82(11): 2191– 2203.
28. Taxy JB, Battifora H, Trujillo Y, Dorfman HD. Electron microscopy in the di agnosis of malignant schwannoma. Cancer 1981; 48(6): 1381– 1391.
29. Perrin RG, Guha A. Malignant peripheral nerve she ath tumors. Ne urosurg Clin N Am 2004; 15(2): 203– 216.
30. Meis JM, Enziger FM, Martz KL, Ne al JA. Malignant peripheral nerve she ath tumors (Malignant schwannomas) in children. Am J Surg Pathol 1992; 16(7): 694– 707.
31. Wanebo JE, Malik JM, VandenBerg SR, Wanebo HJ, Dri esen N, Persing JA. Malignant peripheral nerve she ath tumors. A clinicopathological study of 28 cases. Cancer 1993; 71(4): 1247– 1253.
32. Vege DS, Chinoy RF, Ganesh B, Parikh DM. Malignant peripheral nerve she ath tumors of the he ad and neck: A clinicopathological study. J Surg Oncol 1994; 55(2): 100– 103.
33. Wo odruff JM. Pathology of tumors of the perifpheral nerve she ath in type 1 ne urofibromatosis. Am J Med Genet 1999; 89(1): 23– 30.
34. Das Gupta TK, Brasfi eld RD. Solitary malignant schwannoma. Ann Surg 1970; 171(3): 419– 428.
35. Kim DH, Murovic JA, Ti el RL, Moes G, Kline DG. A seri es of 397 peripheral ne ural she ath tumors: 30- ye ar experi ence at Lo uisi ana State University He alth Sci ences Center. J Ne urosurg 2005; 102(2): 246– 255.
36. Bojsen- Møller M, Myhre‑Jensen O. A consecutive seri es of 30 malignant schwannomas. Survival in relati on to clinico- pathological parameters and tre atment. Acta Path Microbi ol Immunol Scand (A) 1984; 92(3): 147– 155.
37. Storm FK, Eilber FR, Mirra J, Morton DL. Ne urofibrosarcoma. Cancer 1980; 45(1): 126– 129.
38. Foley KM, Wo odruff JM, Ellis FT, Posner JB. Radi ati on‑induced malignant and atypical peripheral nerve she ath tumors. Ann Ne urol 1980; 7(4): 311– 318.
39. Fein DA, Lee WR, Lanci ano RM, Corn BW, Herbert SH, Hanlon AL et al. Management of extremity soft tissue sarcomas with limb- sparing surgery and postoperative irradi ati on: do total dose, overall tre atment time, and the surgery- radi otherapy interval impact on local control? Int J Radi at Oncol Bi ol Phys 1995; 32(4): 969– 976.
40. Cashen DV, Parisi en RC, Raskin K, Hornicek FJ, Gebhardt MC, Mankin HJ. Survival data for pati ents with malignant schwannoma. Clin Orthop Rel Res 2004; 426: 69– 73.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2009 Číslo 2
Nejčtenější v tomto čísle
- Krční myelopatie – diagnostický problém
- Neurodegenerativní demence
- Maligní tumor z pochvy periferního nervu – dvě kazuistiky
- Radi ofrekvenční terapi e facetových bolestí bederní páteře