
Diferenciální diagnostika neuroakantocytóz
Differential Diagnosis of Neuroacanthocytosis
Acanthocytes are atypical erythrocytes with sharp membrane protrusions. The range of diseases classified as neuroacanthocytosis (NA) includes a variety of disorders characterised by the occurrence of acanthocytes together with neurological symptoms. The detection of acanthocytes is, however, not specific and may not necessarily be a symptom of NA. The text describes both the most frequent and rare forms of NA, their etiology, and clinical and laboratory picture. The authors propose their own differential diagnostic approach based on the incidence of atrophy of the caput nuclei caudati which can make diagnosis faster and cheaper.
Key words:
acanthocytes – blood smear – neuroacanthocytosis – chorea – atrophy of caput nuclei caudati – choreoacanthocytosis – McLeod’s syndrome – abetalipoproteinemia
Autoři:
J. Klempíř 1; D. Mikulenková 2; M. Písačka 2; O. Klempířová 1
Působiště autorů:
Neurologická klinika 1. LF UK a VFN, Praha
1; Ústav klinické a experimentální hematologie 1. LF UK a ÚHKT Praha
2
Vyšlo v časopise:
Cesk Slov Neurol N 2009; 72/105(1): 24-29
Kategorie:
Přehledný referát
Souhrn
Akantocyty jsou atypické erytrocyty s ostrými výběžky membrány. Jako neuroakantocytózy (NA) jsou označována velmi různorodá onemocnění, u kterých neurologická symptomatika je doprovázena výskytem akantocytů. Záchyt akantocytů však není specifický a nemusí být obligátním příznakem NA. V textu jsou popsány svým výskytem nejčastější i raritní NA, jejich etiologie, klinický a laboratorní obraz. Autoři navrhují vlastní diferenciálně diagnostický postup, založený na výskytu atrofie caput nuclei caudati, který může stanovení diagnózy urychlit a zlevnit.
Klíčová slova:
akantocyty – krevní nátěr – neuroakantocytóza – chorea – atrofie caput ncl. caudati – choreoakantocytóza – McLeodův syndrom – abetalipoproteinemie
Úvod
Akantocyty jsou atypické erytrocyty s ostrými výběžky membrány (obr. 1). Za jednu z příčin vzniku akantocytů je považována porucha erytrocytární membrány [1,2]. Akantocyty vznikají v periferní krvi, neboť nebyly pozorovány v kostní dřeni. Akantocyty mají zkrácený biologický poločas a snáze podléhají hemolýze.
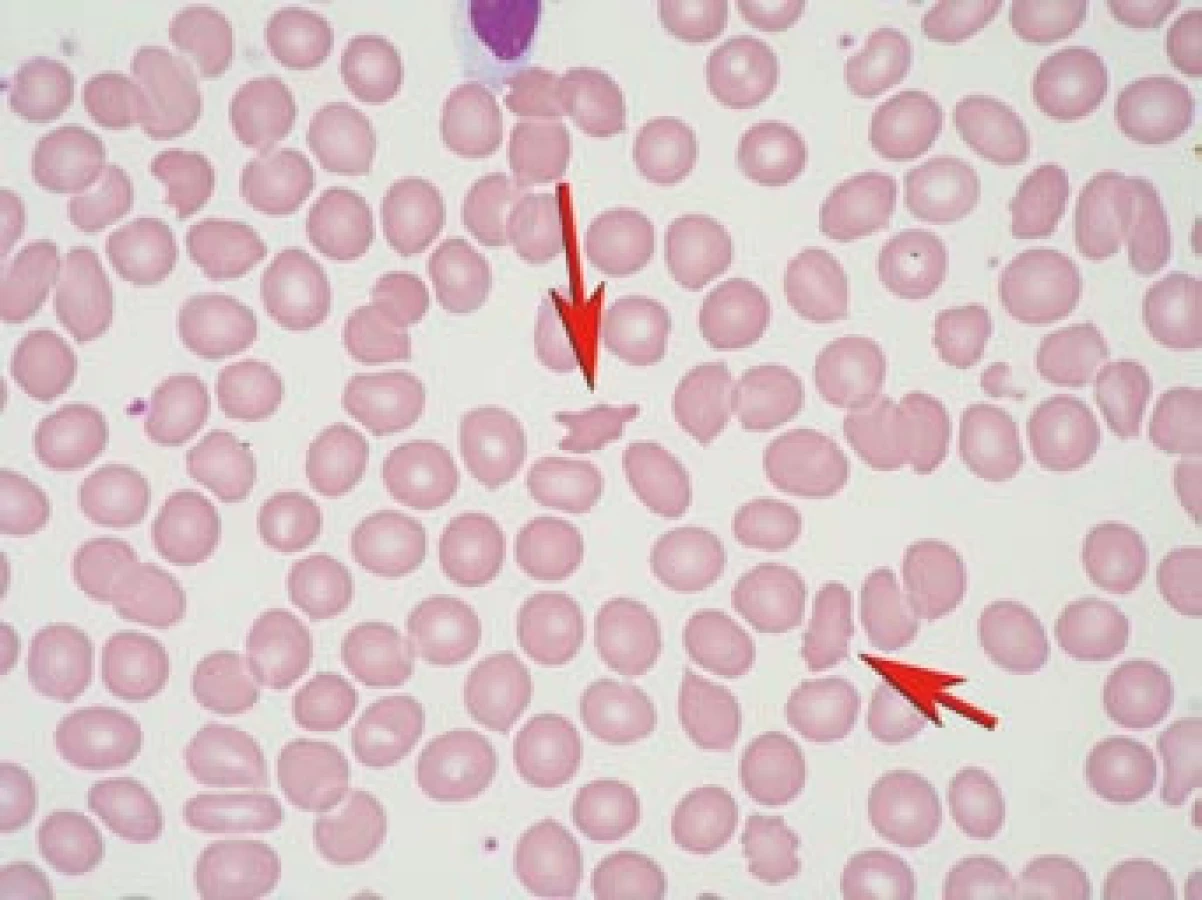
Výskyt akantocytů může být spojen s řadou různých onemocnění (tab. 1). Akantocyty mohou být zachyceny i zcela náhodně v malém počtu bez doprovodné symptomatiky u zdravých osob [3]. Jako první výskyt akantocytů v asociaci s neurologickou symptomatikou (ataxie a pigmentová retinopatie) popsali Bassen a Kornzweig v roce 1950 [4] u abetalipoproteinemie. Pravděpodobně neexistuje kauzální vztah nebo závislost mezi akantocyty a neurologickým postižením.
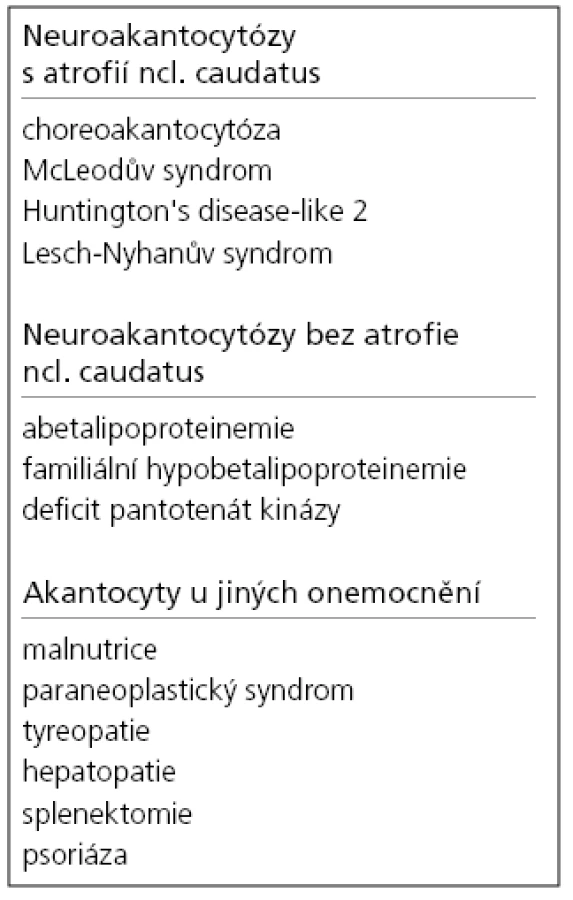
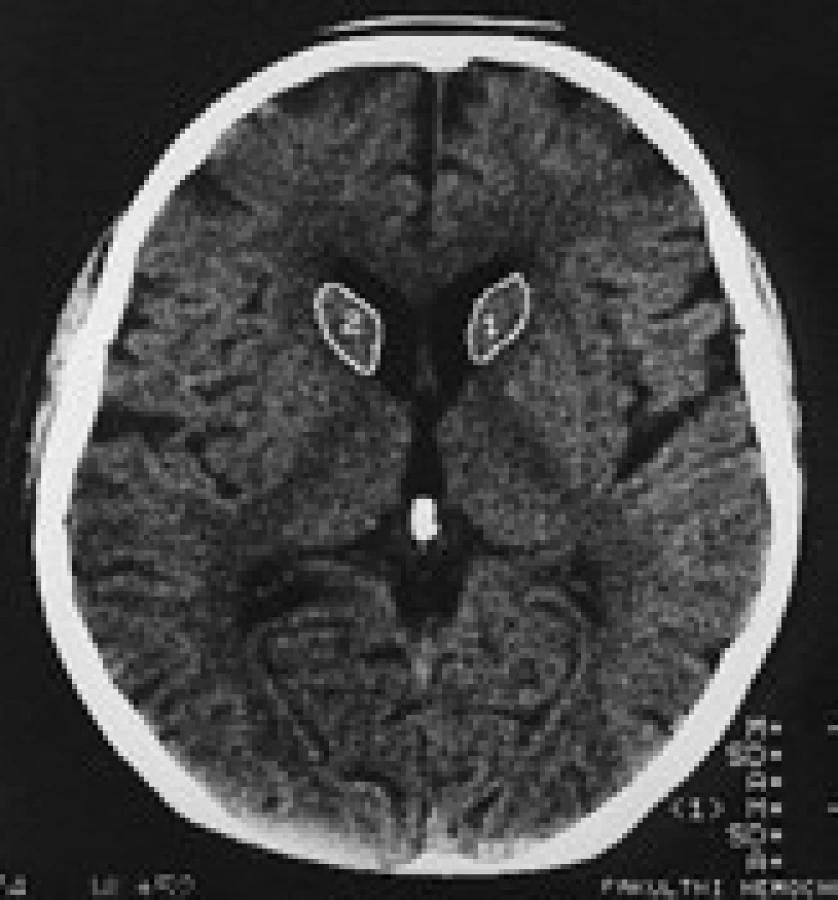
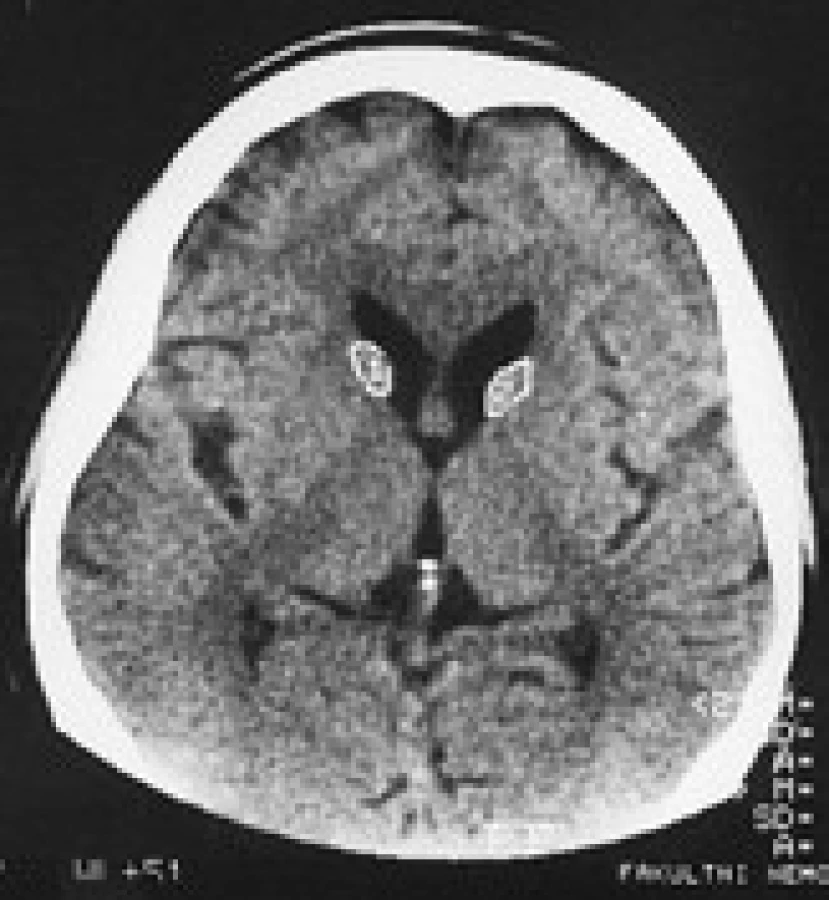
Termín neuroakantocytóza (NA) je v současnosti vyhrazen pro asociaci výskytu akantocytů s neurologickou symptomatikou. NA syndromy jsou vzácné, velmi pestré, různé etiologie, a proto je jejich dělení problematické. Je nutno mít na paměti, že akantocyty u NA nemusí být vždy zachyceny. Danek et al [1] rozdělují NA na dvě základní skupiny: 1. s poruchou metabolizmu lipidů, které postihují zejména míchu, periferní nervový systém a retinu; 2. s dominantním postižením mozku, zejména bazálních ganglií. Diferenciální diagnostiku může urychlit znalost výskytu nejčastějších příčin, typický věk počátku a eventuální familiární způsob přenosu NA syndromů. Některé NA syndromy lze vyloučit genetickým testováním, ale jedná se o drahá a špatně dostupná vyšetření. V našem článku uvádíme dělení podle výskytu atrofie caput nuclei caudati (CNC) (tab. 1, obr. 2, 3), které vychází z naší skromné klinické [5] a literární zkušenosti.
Neuroakantocytózy s atrofií ncl. caudatus
Choreoakantocytóza (Levineův-Critchleyův syndrom, dříve neuroakantocytóza, ChAc) [6,7] je autozomálně recesivně (AR) dědičné onemocnění vázané na 9. chromozom [8] a již bylo popsáno více než 80 mutací. Produktem mutovaného genu je abnormní protein chorein s nejasnou funkcí vyskytující se zejména v tkáních odpovědných za klinický obraz (prekurzory erytrocytů, kosterní sval, mozek) [9]. ChAc se klinicky manifestuje mezi 8.–62. rokem, nejčastěji ve 3. dekádě [1]. Byl však dokumentován počátek ChAc v 86 letech u ženy s negativní rodinnou anamnézou [10]. Pro ChAc je typická kombinace výskytu dyskinetického syndromu a nápadné makroskopické atrofie CNC. Chorea a dystonie se vyskytují u více než poloviny případů. Pro ChAc jsou typické orofaciální dyskinézy včetně automutilace rtů a jazyka, dysartrie, dysfagie, poruch dýchání a vokální tiky. V pokročilých stadiích se objevuje tzv. feeding dystonie (jazyk vytlačuje potravu z úst). Typická je bizarní chůze s trupovou dystonií, která může vést k častým pádům. Ekvinózní postavení nohou může být způsobeno dystonií či oslabením peroneálního svalstva. S progresí ChAc se postupně objevuje a u jedné třetiny nemocných dominuje parkinsonský syndrom [1,11]. Až na výjimky je přítomna šlachově okosticová areflexie a neuropatické změny, které korespondují s neurografickým vyšetřením. Změny ve svalové biopsii jsou však vzácné, přestože je často přítomna svalová atrofie, snížení svalové síly a zvýšená kreatinkináza [12]. V séru je často zvýšena laktátdehydrogenáza, alaninaminotransferáza (ALT), aspartátaminotransferáza (AST), někdy i gamaglutamyltransferáza (GMT) a vždy snížený haptoglobin (marker hemolýzy akantocytů). Přibližně polovina nemocných trpí častými epileptickými záchvaty, kognitivní deteriorací subkortikálního typu a behaviorálními poruchami (viz níže). Rychlost a průběh onemocnění mohou být individuální a již po několika letech těžce invalidizující [12,13].
Ještě vzácnější je X recesivně vázaný McLeodův syndrom (MLS) [14,15], u kterého již bylo popsáno nejméně 15 mutací [16]. Mutace genu XK vede ke snížení tvorby Kell antigenů systému krevních skupin lokalizovaných na erytrocytární membráně [14,15,17,18]. MLS se manifestuje u mužů, mezi 18.–61. rokem, nejčastěji okolo 35. roku [1], a může být klinicky identický s ChAc. Pro MLS je typická kongestivní či dilatační kardiomyopatie a poruchy srdečního rytmu (fibrilace síní, tachyarytmie), hepatosplenomegalie, myopatické změny ve svalové biopsii [19]. Poruchy hlubokého čití jsou častější než u ChAc. Hladina haptoglobinu může být normální. Vzácně se u heterozygotních přenašeček mohou objevit některé příznaky MLS [11,16]. Makroskopicky významná atrofie CNC byla zachycena i u jedince klinicky v presymptomatickém stadiu [16]. V roce 2005 se nám podařilo geneticky verifikovat první případ MLS v České republice [5].
Huntington‘s disease like 2 (HDL2) [20–22] je autozomálně dominantně (AD) dědičná neuroakantocytóza, která byla dosud popsána jen v jedné rozsáhlé mexické rodině, jejíž předci pocházeli z Afriky [22,23]. HDL2 je způsobena nerozštěpením CTG/CAG trinukleotidů řetězce genu pro junctophilin 3 na chromozomu 16q [24]. Klinicky se HDL2 manifestuje ve 2. a 3. dekádě života abulií, depresí, poruchami osobnosti a demencí. Z neurologických příznaků se vyskytuje chorea, dystonie a parkinsonský syndrom, který může v pozdních stadiích dominovat. Rovněž jsou přítomny poruchy polykání pro orolingvální dystonii. U HDL2 nebylo zaznamenáno neuromuskulární postižení. U HDL2 je rovněž výrazná atrofie CNC a také putamen. V několika případech HDL2 byla pro svůj klinický obraz a současný výskyt akantocytů mylně považována za ChAc [25].
Lesch Nyhanův syndrom (LNS) je X vázaná neuroakantocytóza způsobená mutací genu pro enzym hypoxantin guanin fosforibosyltransferázu (HPRT) [26], který katalyzuje recyklaci purinových bazí hypoxantinu a guaninu. Kompletní deficit HPRT má za následek nejen jejich přímou degradaci na kyselinu močovou, ale také jejich zvýšenou kompenzační syntézu. Hyperurikemie vede k ukládání krystalů natrium urátu (urátové tofy) do kostí (na rtg subchondrálně cystická projasnění) a měkkých tkání (ušní boltec, šlachy, kloubní chrupavka, synoviální výstelka) a následné zánětlivé reakci. LNS se manifestuje u chlapců nejčastěji v 1. roce života choreoatetózou, spasticitou, mentální retardací a ve 2. roce automutilací (pokousání rtů a prstů). Současně je přítomna megaloblastická anémie, nefrolitiáza, atrofie varlat a ledvin. Rovněž u LNS je zřejmá atrofie CNC. Většina dětí umírá na renální selhání ve třech letech.
Neurodegenerativní onemocnění s atrofií CNC bez výskytu akantocytů
Nápadná makroskopická atrofie CNC v kombinaci s dyskinetickým syndromem, kognitivním deficitem a behaviorálními poruchami (viz dále) bez záchytu akantocytů se nejčastěji vyskytuje u Huntingtonovy nemoci (HN) [27–29]. Pokud je genetickým vyšetřením HN spolehlivě vyloučena, je nutno zvažovat možnost některého jiného vzácného onemocnění. V zahraniční literatuře je často zdůrazňována nutnost vyloučit také dentato-rubro palido-luisiánskou atrofii a spinocerebellární ataxii typu 3 a typu 17. U těchto onemocnění se však výrazná atrofie CNC, která je u HN [30] a neuroakantocytóz [1] patrná již při klinickém počátku, nevyskytuje [31]. Navíc ataxie nepatří k hlavním příznakům HN či NA. Atrofie CNC a klinický projev připomínající HN byla popsána v několika kazuistikách u AR Huntington's disease like 3 [32] a Huntington‘s disease like 4 [33]. Huntington‘s disease like 3 se projevuje kombinací dyskinetického syndromu, spasticity, ataxie, epileptických záchvatů a demence. U Huntington‘s disease like 4 byly popsána chorea, tremor, rigidita a poruchy chůze.
Neuroakantocytózy bez atrofie ncl. caudatus
Abetaliporoteinemie (Bassen Kornzweig syndrom, ABL) [4] je AR onemocnění způsobené mutací genu pro mikrozomální transportní protein na 4. chromozomu [34]. U ABL chybí apolipoprotein B, který je součástí lipoproteinových částic (chylomikrony, lipoproteiny o velmi nízké hustotě (VLDL) a lipoproteiny o nízké hustotě (LDL)). Chybění VLDL a LDL vede k velmi nízké hladině triglyceridů a cholesterolu [35,36]. Sekundární malabsorbce tuků v tenkém střevě se často projeví již v prvním roce života steatoreou a distenzí střev, která vede k podezření na celiakální sprue. Mentální retardace se objevuje u třetiny nemocných. Následně bývá přítomna porucha růstu, v pátém roce šlachově okosticová areflexie, v 10. roce senzorická neuropatie, ataxie, tremor a oslabení svalové síly. U ABL se objevuje nystagmus, poruchy okulomotoriky (oftalmoplegie) a poruchy vizu (retinitis pigmentóza, šeroslepost, koncentrické zúžení zorného pole a úbytek zrakové ostrosti). Neurologická symptomatika je výsledkem deficitu vitaminu E, který podmiňuje mimo jiné i úbytek postsynaptických dopaminových receptorů, pravděpodobně pro zvýšený oxidativní stres [37], ale ne makroskopickou atrofii bazálních ganglií. Sníženy jsou i hladiny ostatních liposolubilních vitaminů. Deficit vitaminu K může vést k hemoragickým příhodám. Bez substituce chybějících vitaminů nemocní umírají okolo 40. roku života [38].
Podobným mechanizmem jako ABL je způsobena AD familiární hypoprebetalipoproteinemie s odlišnou mutací apolipoproteinu B [39,40]. Klinický obraz může být podobný ABL, ale neurologické postižení je vyjádřeno méně [35,41]. Heterozygoti jsou obvykle asymptomatičtí, nemají akantocyty, ale sérové hladiny TAG, LDL a VLDL jsou významně sníženy. U homozygotů je přítomna steatorea, v dospělosti se objevuje ataxie, neuropatie, retinální degenerace a akantocyty. Mezi další onemocnění s poruchou metabolizmu lipidů často sdružených s neurologickou symptomatikou patří AR onemocnění s retencí chylomikronů [42]. Podobný klinický obraz bez akantocytózy může být i u AR onemocnění, jako jsou deficit vitaminu E a Friedreichova ataxie [43].
Deficit pantotenát kinázy (dříve označovaný za Hallervorden-Spatzův syndrom, PKAN) se převážně objevuje v dětství, vzácně v adolescenci a počátek v dospělosti je raritou [44]. PKAN je AR onemocnění s mutací genu pro pantotenát kinázu 2 na 20. chromozomu [45]. Pro PKAN je typická obličejová dystonie, dystonické držení trupu, poruchy chůze, posturální instabilita a pigmentová degenerace retiny. Může se objevit choreoatetóza, hyperextenze prstů, pes cavus či ekvinózní postavení nohou. U PKAN mohou být zachyceny akantocyty [46]. Kognitivní deficit subkortikálního typu (viz dále) patří do obrazu PKAN, ale většinou nedospěje do stadia demence. Syndrom HARP (hypoprebetalipoproteinemie, akantocytóza, retinitis pigmentosa a palidální degenerace) se ukázal jako alelická varianta PKAN [47–49].
Akantocyty se mohou vyskytovat i u jiných systémových onemocnění v kombinaci s neurologickou symptomatikou [12,45,50]. Při těžkých malnutričních stavech (mentální anorexie, nádorová kachexie) se mohou objevit akantocyty. Akantocyty mohou být zachyceny v rámci jaterní cirhózy s encefalopatií, uremie, paraneoplastického syndromu, psoriázy a tyreopatie. Akantocyty se objevují i po splenektomii, neboť ve slezině dochází k degradaci erytrocytů [51].
NA v CT a MR obraze
V CT a MR obraze je makroskopická atrofie podobně jako u HN [27,30] i u ChAC a MLS nejvíce patrná v CNC [16]. Výrazná atrofie CNC je patrná již na počátku klinických obtíží. Na našem pracovišti se osvědčila CT planimetrie CNC (obr. 2, 3) [27]. V MR obrazech je vidět i atrofie putamen a globus pallidus, která je výraznější u ChAC než u MLS. U ChAc lze stejně jako u HN v MR T2W obrazech pozorovat nespecifické zvýšení signálu ve striatu. Pro HDL2 je typická masivní globální atrofie mozkové kůry a atrofický gradient striata podobný jako u HN [21]. Pro PKAN je v MR T2W obrazech příznačný pokles signálu v globus pallidus a s centrální hyperintenzitou (obraz tygřích očí) [52]. Nápadná atrofie mozkového kmene a mozečku není pro NA typická.
Kognitivní a behaviorální projevy NA
Degenerativní procesy v bazálních gangliích jsou většinou spojeny s kognitivním deficitem a behaviorálními změnami. Bazální ganglia jsou propojena s frontální kůrou i limbickým systémem. Z toho vyplývá podobný charakter psychického postižení u těchto onemocnění. Obvykle se nejprve objevují izolované poruchy pozornosti, učení, pracovní paměti, okamžitého vybavení z paměti, vizuálně percepční deficity a poruchy exekutivních funkcí. Exekutivní funkce slouží k efektivní tvorbě a realizaci plánů, analogií, vykonávání paralelních kognitivních úloh a adaptaci na nové události. Globální kognitivní výkon zůstává často dlouho v normě, ale s progresí nemoci se kognitivní deficity sdružují a dosahují míry demence tzv. subkortikálního typu s dominantním postižením exekutivních funkcí [5–7,16,29,40]. Behaviorální poruchy se manifestují nejčastěji apatií, úzkostí a depresí. Objevuje se i psychomotorický neklid, impulzivita, iritabilita, obsedantně kompulzivní poruchy, tikové poruchy a vzácně psychotické příznaky [5–7,16, 29,40]. Izolované kognitivní deficity a poruchy chování mohou předcházet neurologickému postižení bazálních ganglií řadu let [53,54].
Laboratorní vyšetření
Elevace jaterních parametrů může být v rámci ChAc, MLS, Wilsonovy nemoci, abúzu alkoholu či jiného chronického jaterního postižení. Vhodné je sonografické vyšetření jater a sleziny. Pokud není vyjádřena nápadná atrofie CNC, je žádoucí spolehlivě vyloučit Wilsonovu nemoc (odpad mědi v moči a Kayser-Fleischerův prstenec nejsou spolehlivými ukazateli, zlatým standardem je množství mědi v jaterní sušině anebo pozitivní průkaz genetické mutace). Příčiny a význam zvýšené CK a LDH u ChAc a MLS nejsou jasné. Snížení cholesterolu a triglyceridů či změny v elektroforetickém spektru lipidů ukazují na možnost malabsorpce lipidů (ABL, familární hypoprebetalipoproteinemii, HARP). Vhodné je vyšetřit hladinu vitaminu E a funkci štítné žlázy.
Kardiologické vyšetření by mělo být zaměřeno na výskyt arytmií a kardiomyopatie. Při klinických známkách neuropatie je dobré provést neurografické vyšetření a jehlové elektromyografické vyšetření. Oftalmolog by měl sledovat poruchy okulomotoriky a degenerativní procesy na sítnici.
Hematologické vyšetření akantocytóz
Před speciálním hematologickým vyšetřením doporučujeme provést sedimentaci, krevní obraz a diferenciální rozpočet. Morfologicky jsou akantocyty definovány jako atypické erytrocyty přibližně sférického tvaru, které mají na svém povrchu nepravidelně distribuované výběžky nestejné délky i tvaru. Nejčastěji mají ostré výběžky, mohou ale mít i tvar paličky. V minimálním počtu (do 0,5%) lze jejich přítomnost v nátěru periferní krve barveném postupem May-Grünwald-Giemsa (MGG) považovat za nepatologickou. K vyloučení záměny s echinocyty v panoptickém MGG barvení je lepší nátěr krve vyšetřit ve fázovém kontrastu či elektronovým mikroskopem. Zvýšená přítomnost akantocytů, tj. akantocytóza, doprovází řadu vrozených i získaných onemocnění s neurologickou symptomatologií i bez ní. Akantocyty ale nejsou obligátním příznakem neuroakantocytóz. Jejich nepřítomnost v panoptickém barvení nevylučuje toto onemocnění a jejich množství též nesouvisí s tíží neurologické choroby. Dosud neexistuje standardizovaná metoda k vyšetření přítomnosti akantocytů, ale lze doporučit postup německých neurologů [3], kteří u 137 neurologických pacientů a 37 dobrovolníků použili tři typy vzorků: 5ml krve s EDTA, krev bez antikoagulačního činidla smíchaném ve stejném množství izotonického fyziologického roztoku a krev po smíchání s levomepromazinem (způsobuje reverzibilitu tvorby echinocytů). Po inkubaci při pokojové teplotě buď suchý nátěr krve obarvili klasicky dle MGG, či mokrý nefixovaný preparát zamontovali s krycím sklem. Vzorky zkoumali v mikroskopu s fázovým kontrastem a v elektronovém mikroskopu. Z výsledků vyplývá doporučení použít k potvrzení neuroakantocytózy vzorek krve s izotonickým roztokem a prohlédnout nefixovaný „mokrý“ preparát. Za patologickou hodnotu je považováno více než 6,3% akantocytů.
Vyšetření antigenů Kell systému se provádí na nativních erytrocytech, nejlépe na čerstvém odběru antikoagulovaného vzorku, přičemž na antikoagulans nezáleží. Tyto antigeny lze vyšetřit i ve vzorku sražené krve, ale vyšetření je o něco komplikovanější a nelze z takového vzorku kvalitně izolovat DNA pro případná následná molekulárněbiologická vyšetření. Spolehlivost sérologické diagnostiky antigenů Kell systému je vysoká. V České republice se provádí rutinní určování antigenů Ka k na erytrocytární membráně certifikovanými diagnostiky na každém větším transfuzním oddělení. Detekce erytrocytárních Kp(a) a Kp(b) antigenů certifikovanými diagnostiky je méně běžné, avšak dostupné vyšetření ve specializovanějších laboratořích transfuzní služby. Kx antigen prokazujeme pouze na našem pracovišti (Referenční laboratoř pro imunohematologii), pomocí vzácného anti Kx séra, které není komerčně dostupné.
Genetická verifikace onemocnění, u nichž se vyskytují akantocyty, je v České republice možná u Lesch-Nyhanova syndromu, abetaliporoteinemie, familiální hypobetalipoproteinemie např. v Ústavu dědičných metabolických poruch 1. LF UK a VFN a deficitu pantotenát kinázy v Ústavu biologie a lékařské genetiky 2. LF UK v Praze. V případě vážného podezření na mutaci XK genu je možné sekvenační vyšetření v Mezinárodní referenční laboratoři pro krevní skupiny v Bristolu prostřednictvím Referenční laboratoře v Ústavu hematologie a krevní transfuze v Praze. Detekce choreinu u ChAc se zatím provádí pouze v Mnichově (http://www.nefo.med.uni-muenchen.de/~adanek/Chorein_Blot.pdf).
Závěr
Výskyt a záchyt akantocytů nemusí být spolehlivým příznakem NA. Pokud je současně s dyskinetickým syndromem, kognitivní deteriorací a behaviorálními poruchami přítomna makroskopická atrofie CNC, je třeba vyloučit nejprve HN. Na možnost jiného onemocnění (ChAc nebo MLS) ukazuje zejména bohatší symptomatika (epilepsie, neuromuskulární postižení, hepatopatie, splenomegalie, elevace CK). Pro MLS je typické mužské pohlaví, kardiomyopatie a deficit tvorby Kell antigenů. HDL2 se vyskytuje jen u jedinců, jejichž rodiče pocházeli z Afriky. Lesch Nyhanův syndrom se manifestuje v prvních letech života. U NA bez atrofie CNC je třeba vyloučit poruchy metabolizmu lipidů bez dyskinezí (ABL a familiární hypoprebetalipoproteinemie) a s dyskinetickým syndromem (HARP syndrom), která bývají spojeny s pigmentovou degenerací retiny. Negativní rodinná anamnéza nevylučuje žádné z výše jmenovaných onemocnění.
Práce byla podpořena grantem IGA MZ ČR NR8937-4 a Výzkumným záměrem MSM0021620849.
MUDr. Jiří Klempíř, Ph.D.
Centrum extrapyramidových
onemocnění
Ne urologická klinika
1. LF UK a VFN
Kateřinská 30
128 08 Praha
e‑mail: jiri.klempir@seznam.cz
Zdroje
1. Danek A, Jung HH, Melone MAB, Rampoldi L, Broccoli V, Walker RH. Neuroacanthocytosis: new developments in a neglected group of dementing disorders. J Neurol Sci 2005; 229–230: 171–186.
2. De Franceschi L, Olivieri O, Corrocher R. Erythrocyte aging in neurodegenerative disorders. Cell Mol Biol 2004; 50(2): 179–185.
3. Storch A, Kornhass M, Schwarz J. Testing for acanthocytosis A prospective reader-blinded study in movement disorder patients. J Neurol 2005; 252(1): 84–90.
4. Bassen FA, Kornzweig AL. Malformation of the erythrocytes in a case of atypical retinitis pigmentosa. Blood 1950; 5(4): 381–387.
5. Klempír J, Roth J, Zárubová K, Písacka M, Spacková N,Tilley L. The McLeod syndrome without acanthocytes. Parkinsonism Relat Disord 2008; 14(4): 364–366.
6. Levine IM, Estes JW, Looney JB. Hereditary neurological disease with acanthocytosis, a new syndrome. Arch Neurol 1968; 19(4): 403–409.
7. Critchley EM, Clark DB, Wikler A. Acanthocytosis and neurological disorder without betalipoproteinemia. Arch Neurol 1968; 18(2): 134–140.
8. Rubio JP, Danek A, Stone C, Chalmers R, Wood N, Verellen C et al. Chorea-acanthocytosis: genetic linkage to chromosome 9q21. Am J Hum Genet 1997; 61(4): 899–908.
9. Ueno S, Maruki Y, Nakamura M, Tomemori Y, Kamae K,Tanabe H et al. The gene encoding a newly discovered protein, chorein, is mutated in chorea-acanthocytosis. Nat Genet 2001; 28(2): 121–122.
10. Ikawa M, Yoneda M, Kuriyama M. A case of chorea-acanthocytosis onset with at age 86. Rinsho Shinkeigaku. 2005; 45(8): 603–606.
11. Hardie RJ, Pullon HW, Harding AE, Owen JS, Pires M,Daniels GL et al. Neuroacanthocytosis: a clinical, haematological and pathological study of 19 cases. Brain 1991; 114(1A): 13–49.
12. Alonso ME, Teixeira F, Jimenez G, Escobar A. Chorea-acanthocytosis: report of a family and neuropathological study of two cases. Can J Neurol Sci 1989; 16(4): 426–431.
12. Stevenson VL, Hardie RJ. Acanthocytosis and neurological disorders. J Neurol 2001; 248(2): 87–94.
13. Aasly J, Skandsen T, Rø M. Neuroacanthocytosis – the variability of presenting symptoms in two siblings. Acta Neurol Scand 1999; 100(5): 322–325.
14. Allen FH jr, Krabbe SM, Corcoran PA. A new phenotype (McLeod) in the Kell blood–group system. Vox Sang 1961; 6: 555–560.
15. Marsh WL, Marsh NJ, Moore A, Symmans WA, Johnson CL, Redman CM. Elevated serum creatine phosphokinase in subjects with McLeod syndrome. Vox Sang 1981; 40(6): 403–411.
16. Danek A, Rubio JP, Rampoldi L, Ho MF, Dobson-Stone C, Tison F et al. McLeod neuroacanthocytosis: genotype and phenotype. Ann Neurol 2001; 50(6): 755–764.
17. Wimer BM, Marsh WL, Taswell HF, Galey WR. Haematological changes associated with the McLeod phenotype of the Kell blood group system. Br J Haematol 1977; 36(2): 219–224.
18. Russo D, Redman C, Lee S. Association of XK and Kell blood group proteins. J Biol Chem 1998; 273(22): 13950–13956.
19. Jung HH, Hergersberg M, Vogt M, Pahnke J, Treyer V,Rothlisberger B et al. McLeod phenotype associated with a XK missense mutation without hematologic, neuromuscular, or cerebral involvement. Transfusion 2003; 43(7): 928–938.
20. Margolis RL, O’Hearn E, Rosenblatt A, Willour V, Holmes SE, Franz ML et al. A disorder similar to Huntington’s disease is associated with a novel CAG repeat expansion. Ann Neurol 2001; 50(6): 373–380.
21. Walker RH, Morgello S, Davidoff-Feldman B, Melnick A, Walsh P, Shashidharan P et al. Autosomal dominant chorea-acanthocytosis with polyglutamine-containing neuronal inclusions. Neurology 2002; 58(7): 1031–1037.
22. Walker RH, Jankovic J, O’Hearn E, Margolis RL. Phenotypic features of Huntington’s disease like 2. Mov Disord 2003; 18(12): 1527–1530.
23. Stevanin G, Camuzat A, Holmes SE, Julien C, Sahloul R, Dode C et al. CAG/CTG repeat expansions at the Huntington’s disease like 2 locus are rare in Huntington’s disease patients. Neurology 2002; 58(6): 965–967.
24. Holmes SE, O’Hearn E, Rosenblatt A, Callahan C,Hwang HS, Ingersoll-Ashworth RG et al. A repeat expansion in the gene encoding junctophilin 3 is associated with Huntington disease like 2. Nat Genet 2001; 29(4): 377–378.
25. Walker RH, Rasmussen A, Rudnicki D, Holmes SE, Alonso E, Matsuura T et al. Huntington‘s disease – like 2 can present as chorea-acanthocytosis. Neurology 2003; 61(7): 1002–1004.
26. Jolly DJ, Esty AC, Bernard HU, Friedmann T. Isolation of a genomic clone partially encoding human hypoxanthine phosphoribosyltransferase. Proc Natl Acad Sci USA 1982; 79(16): 5038–5041.
27. Roth J, Klempíř J, Jech R, Židovská J, Uhrová T, Doubek P et al. The caudate nucleus atrophy in Huntington’s disease and its relation to clinical and genetic parameters. Func Neurol 2005; 20(3):127–130.
28. Roth J, Klempíř J, Špačková N. Neuropsychologie Huntingtonovy nemoci. In: Preiss M, Kučerová O (eds). Neuropsychologie pro neurology. Praha: Grada 2006.
29. Klempíř J, Klempířová O, Roth J. Bazální ganglia a paměť. In: Hort J, Rusina R (eds). Paměť a její poruchy. Praha: Maxdorf 2008: 330–335.
30. Aylward EH, Sparks BF, Field KM, Yallapragada V, Shpritz BD, Rosenblatt A et al. Onset and rate of striatal atrophy in preclinical Huntington disease. Neurology 2004; 63(1): 66–72.
31. Schöls L, Bauer P, Schmidt T, Schulte T, Riess O. Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. The Lancet 2004; 3(5): 291–304.
32. Al-Tahan AY, Divakaran MP, Kambouris M, Bohlega S, Salih M, Ogunniyi A et al. A novel autosomal recessive ‚Huntington‘s disease like‘ neurodegenerative disorder in a Saudi family. Saudi Med J 1999; 20: 85–89.
33. Richfield EK, Vonsattel JP, MacDonald ME, Sun ZQ, Deng A, Reiner A. Selective loss of striatal preprotachykinin neurons in a phenocopy of Huntington’s disease. Mov Disord 2002; 17(2): 327–332.
34. Narcisi TM, Shoulders CC, Chester SA, Read J, Brett DJ, Harrison GB et al. Mutations of the microsomal triglyceride-transfer-protein gene in abetalipoproteinemia. Am J Hum Genet 1995; 57(6): 1298–1310.
35. Kane JP, Havel R. Metabolic and Molecular Basis of Inherited Disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds). 7th ed. New York: McGraw Hill Publishing Co 1995: 1853–1885.
36. Welty FK, Lahoz C, Tucker KL, Ordovas JM, Wilson PW, Schaefer EJ. Frequency of ApoB and ApoE gene mutations as causes of hypobetalipoproteinemia in the framingham offspring population. Arterioscler Thromb Vasc Biol 1998; 18(11): 1745–1751.
37. Dexter DT, Brooks DJ, Harding AE, Burn DJ, Muller DPR, Goss-Sampson MA et al. Nigrostriatal function in vitamin E deficiency: clinical, experimental, and positron emission tomographic studies. Ann Neurol 1994; 35(3): 298–303.
38. Kayden HJ, Traber MG. Absorption, lipoprotein transport, and regulation of plasma concentrations of vitamin E in humans. J Lipid Res 1993; 34(3): 343–358.
39. Linton MF, Farese RV jr, Young SG. Familial hypobetalipoproteinemia. J Lipid Res 1993; 34(4): 521–541.
40. Rampoldi L, Danek A, Monaco AP. Clinical features and molecular bases of neuroacanthocytosis. J Mol Med 2002; 80(8): 475–491.
41. Brin MF. Acanthocytosis. In: Goetz CG, Tanner CM, Aminoff MJ (eds) Handbook of clinical neurology: Systemic diseases, Part I, vol. 19. Amsterdam: Elsevier 1993: 271–299.
42. Shoulders CC, Naoumova RP. The genes and proteins of atherogenic lipoprotein production. Biochem Soc Trans 2004; 32(1): 70–74.
43. Hammans SR, Kennedy CR. Ataxia with isolated vitamin E deficiency presenting as mutation negative Friedreich‘s ataxia. J Neurol Neurosurg Psychiatry 1998; 64(3): 368–370.
44. Zarranz JJ, Gómez-Esteban JC, Atarés B, Lezcano E,Forcadas M. Tau-predominant associated pathology in a sporadic late-onset Hallervorden-Spatz syndrome. Mov Disord 2006; 21(1): 107–111.
45. Zhou B, Westaway SK, Levinson B, Johnson MA, Gitschier J, Hayflick SJ. A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. Nat Genet 2001; 28(4): 345–349.
46. Swisher CN, Menkes JH, Cancilla PA, Dodge PR. Coexistence of Hallervorden-Spatz disease with acanthocytosis. Trans Am Neurol Ass 1972; 97: 212–216.
47. Higgins JJ, Patterson MC, Papadopoulos NM, Brady RO, Pentchev NW, Barton NW. Hypoprebetalipoproteinemia, acanthocytosis, retinitis pigmentosa, and pallidal degeneration (HARP syndrome). Neurology 1992; 42(1): 194–198.
48. Ching KH, Westaway SK, Gitschier J, Higgins JJ, Hayflick SJ. HARP syndrome is allelic with pantothenate kinase associated neurodegeneration. Neurology 2002; 58(11): 1673–164.
49. Houlden H, Lincoln S, Farrer M, Cleland PG, Hardy J,Orrell RW. Compound heterozygous PANK2 mutations confirm HARP and Hallervorden-Spatz syndromes are allelic. Neurology 2003; 61(10): 1423–1426.
50. Tison F. The differential diagnosis of neuroacanthocytosis: an overview. In: Danek A (ed). Neuroacanthocytosis syndromes. Dordrecht: Springer 2004: 15–20.
51. Biemer JJ. Acanthocytosis-biochemical and physiological considerations. Ann Clin Lab Sci 1980; 10(3): 238–249.
52. Sethi KD, Adams RJ, Loring DW, el Gammal T. Hallervorden-Spatz syndrome: clinical and magnetic resonance correlations. Ann Neurol 1988; 24(5): 692–694.
53. Bruneau MA, Lespérance P, Chouinard S. Schizophrenia like presentation of neuroacanthocytosis. J Neuropsychiatry Clin Neurosci 2003; 15(3): 378–380.
54. Zeman A, Daniels G, Tilley L, Dunn M, Toplis L, Bullock T et al. McLeod Syndrome: long life neuropsychiatric disorder due to a novel mutation XK gene. Psychiatr Genet 2005; 15(4): 291–293.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2009 Číslo 1
Nejčtenější v tomto čísle
- Trombotická trombocytopenická purpura (TTP) u pacientky s roztroušenou sklerózou – kazuistika
- Hereditární neuropatie
- Možnost predikce průběhu herpetické encefalitidy pomocí magnetické rezonance – kazuistika
- Diferenciální diagnostika neuroakantocytóz