
Myozitida s protilátkami proti NXP2
Authors:
J. Junkerová 1,2; M. Sabela 1,2; E. Kovalová 2
Authors‘ workplace:
Katedra a centrum klinických neurověd, LF OU, Ostrava
1; Neurologická klinika FN Ostrava
2
Published in:
Cesk Slov Neurol N 2022; 85(2): 185-186
Category:
Letters to Editor
doi:
https://doi.org/10.48095/cccsnn2022185
Vážená redakce,
idiopatické zánětlivé myopatie, myozitidy, patří standardně do diferenciálně diagnostické rozvahy při subakutním rozvoji svalové slabosti, přestože roční incidence zdokumentovaných případů není vysoká – 11/ 1 000 000 obyvatel, incidence 25/ 100 000 [1]. Z obecné zkušenosti s klinickým obrazem a odezvou na imunosupresivní léčbu výrazně vybočuje případ mladého muže, u kterého jsme diagnostikovali myozitidu s pozitivitou protilátek proti nuclear matrix proteinu 2 (NXP2).
Třiatřicetiletý muž byl neurologem poprvé vyšetřen v listopadu 2020. Stěžoval si na 3 týdny trvající bolesti svalů, nevýkonnost a pozvolna postupující slabost. Jednalo se o doposud zdravého muže a potížím nepředcházely fyzické vypětí, vliv medikamentů či toxinů. Rodinná anamnéza byla stran myopatie negativní. Objektivně byl normální neurologický nález vč. trofiky a síly proximálních i distálních svalů, kůže byla bez exantému. Sérologické testy nevykazovaly abnormity krom signifikantní elevace sérové kreatinkinázy CK-s 21 μkat/ l (referenční rozmezí 0,77–2,85) a myoglobinu 400 μg/ l (referenční rozmezí 0–110). V jehlové EMG vstupně dominovala patologická spontánní aktivita bez typické myogenní přestavby potenciálů motorických jednotek. K rozvoji objektivního oslabení svalové síly proximálních končetinových svalů ke stupni 3 svalového testu došlo během následujících 3 týdnů, souběžně s korelujícím EMG obrazem myopatie. Hladiny CK-s 120 μkat/ l a myoglobinu 2 441 μg/ l svědčily pro rabdomyolýzu bez klinických či laboratorních známek selhání ledvin. V séru byla zjištěna vysoká pozitivita myozitických protilátek anti-NXP2 a hypoalbuminemie bez proteinurie. Klinický obraz bolestivé myopatie se zhoršoval, pacient zaznamenal nárůst hmotnosti o 8 kg, nápadná byla povšechná pastózní tuhost podkoží. Svalová biopsie v 9. týdnu potíží popsala fokální výskyt nekrotických vláken s úklidovou makrofagickou reakcí a disperzní přítomnost T-lymfocytů s imunofenotypem CD4 v intersticiu. Nález potvrdil diagnózu idiopatické zánětlivé myopatie i přes už zavedenou léčbu intravenózními (i.v.) kortikoidy. Desátý týden potíží se projevila dysfagie. Pacient nadále neměl kožní afekce. Následné opakované podání plné dávky i.v. imunoglobulinů (2 g/ kg) vedlo ke snížení hladin CK-s a myoglobinu-s, ale neovlivnilo těžký klinický stav. Do chronické medikace byl zaveden mykofenolát mofetil v dávce 2 g denně. MR svalů provedená v 12. týdnu trvání potíží prokázala výrazný difuzní edém svalů a podkoží (obr. 1), bez změny při kontrole ve 20. týdnu. Kontrolní EMG měla typický myopatický vzorec. PET CT vyloučila malignitu. Nezadržitelná progrese svalové slabosti vedla od 13. týdne onemocnění k imobilitě a nutnosti zavedení perkutánní endoskopické gastrostomie (PEG). Během hospitalizace byla 16. týden zjištěna pozitivita testu PCR na COVID-19. Přes absenci klinických projevů infekce byla vzhledem k imunosupresi zavedena léčba remdesivirem. Spontánní ventilace nebyla ovlivněna postupem choroby ani těmito okolnostmi. Léčebný postup a zvažovaná indikace rituximabu byla opakovaně konzultována s odborníky z Revmatologického ústavu v Praze a vedla k rozhodnutí tam pacienta v březnu 2020 přeložit. Pacient byl léčen tofacitinibem (inhibitorem Janus-kinázy), ale ani tato léčba nevedla ke stabilizaci či zlepšení stavu. Pro aspirační pneumonii a následný rozvoj multiorgánového selhání byl pacient 27. týden přeložen na Kliniku anesteziologie, resuscitace a intenzivní medicíny Fakultní nemocnice Motol. S infaustní prognózou byl následně přeložen na lůžko intenzivní péče v místě bydliště, kde v květnu 2020, 30 týdnů od začátku klinických projevů myozitidy, zemřel. Z důvodů restrikcí během pandemie COVID-19 pitva nebyla provedena.
STIR – short tau inversion recovery
Fig. 1. Thighs MRI from January 1, 2021 (T2 kor 3D space, STIR sequences) – diffuse
edema of muscles and subcutaneous tissue.
STIR – short tau inversion recovery
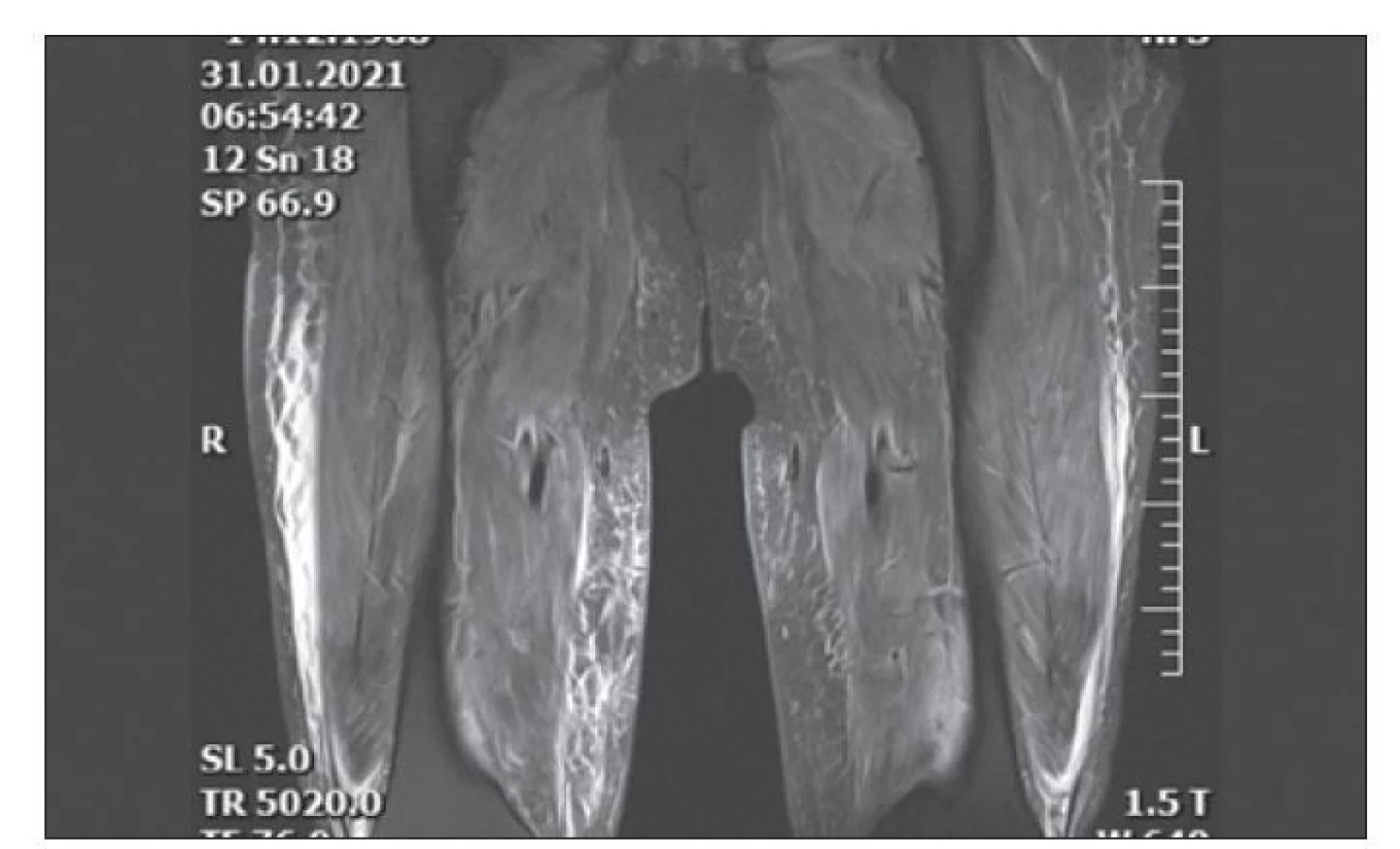
Diagnóza našeho pacienta byla idiopatická zánětlivá myopatie – adermopatická forma dermatomyozitidy s pozitivitou protilátek anti-NXP2.
Termín idiopatické zánětlivé myopatie, myozitidy, zastřešuje více jednotek s podobným klinickým obrazem progredující svalové slabosti, signifikantní elevací CK a myoglobinu v séru a různou mírou kožních či systémových příznaků. Kromě myozitidy s inkluzními tělísky (inclusion body myositis; IBM) klinik ani elektromyografista nerozezná tyto etiopatogeneticky unikátní typy. Stěžejní diagnostickými metodami jsou svalová biopsie a průkaz patologických protilátek v séru [2,3].
Typ protilátek a histopatologický nález u našeho pacienta vedl k diagnóze dermatomyozitidy (DM), přestože kožní projevy nemoci nebyly přítomny. Jde o raritní, tzv. adermopatický typ DM, pro který je asociace s protilátkami anti-NXP2 typická. V literatuře je tento typ označován i jako DMSD (dermatomyozitis sine dermatitis) [4].
Protilátky proti NXP2 jsou známy od roku 1997, ale až v roce 2007 jim byl přiřazen klinický a histopatologický obraz juvenilní DM s převažující či výlučně svalovou symptomatikou [3]. Formy s protilátkami anti-NXP2 tvoří 6 % idiopatických zánětlivých myopatií a 12 % DM. U juvenilních DM se protilátky anti-NXP2 vyskytují v 25 %, u adultních DM pouze v 10 % [4].
Klinicky jsou vždy přítomny těžké projevy myopatie s výraznou elevací CK a myoglobinu v séru. Bolestivá svalová slabost postihuje všechny svalové skupiny končetin, trupu a polykací svaly. Právě dysfagie, rozvinutá časně a intenzivněji než u ostatních forem, je pro anti-NXP2 typ DM typická, vyskytuje se u 74 % případů [4]. Příčně pruhované svaly vč. svalů horní etáže jícnu, hladká svalovina střev a podkoží celého těla jsou výrazně edematózní, což způsobuje nárůst tělesné hmotnosti a redistribuci extracelulární a intracelulární tekutiny s obrazem hypoalbuminemie. Tyto změny lze velmi dobře zobrazit MR svalů, přičemž u anti-NXP2 negativních forem DM se tento obraz nevyskytuje [6]. Může se projevit i kalcinóza podkoží. Není vyšší výskyt intersticiálních plicních procesů, koincidenci s malignitami literární práce potvrzují [7,8], ale i popírají [9]. Anti- NXP2 typ DM má rychlejší progresi, těžkou intenzitu svalové slabosti, polycyklický průběh a horší prognózu, způsobenou nedostatečnou odpovědí na imunosupresivní léčebu [9]. Mortalita této raritní formy DM není stanovena, kazuistiky ale vždy referují o těžkém průběhu a nutnosti využít kombinovanou imunosupresivní léčbu či nestandardní léčebné postupy. Léčba této i jiných refrakterních forem idiopatických zánětlivých myopatií by měla probíhat ve specializovaných centrech. Revmatolog je stěžejní součástí léčebného týmu – rozhoduje např. o využití inhibitorů Janus-kinázy v případě, že všechny jiné léčebné postupy nejsou dostatečně efektivní.
Případ našeho pacienta byl poučný. Pro stanovení správné diagnózy byl stěžejní průkaz silné pozitivity specifických protilátek v séru a histopatologický nález ze svalové biopsie. Naše klinická zkušenost se shoduje s literárními poznatky o raritní anti- NXP2 formě DM – šlo o mladého muže, rychle progredující svalová slabost postihla kosterní i polykací svaly a vedla k imobilitě, byl přítomen masivní otok svalů a podkoží s nárůstem tělesné hmotnosti, nevyskytly se kožní příznaky, jako je heliotropní exantém a rash. Přes intenzivní imunosupresivní léčbu se nepodařilo zvrátit ani stabilizovat průběh choroby. Asymptomatická pozitivita onemocnění COVID-19 průběh choroby neovlivnila. Komplikace velmi těžkého klinického stavu v rámci diagnózy anti-NXP2 formy DM vedly k úmrtí 8 měsíců od začátku potíží.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Přijato k recenzi: 18. 1. 2022
Přijato do tisku: 16. 3. 2022
MUDr. Jana Junkerová
Neurologická klinika
LF OU a FN Ostrava
17. listopadu 1790/5
708 00 Ostrava-Poruba
e-mail: jana.junkerova@fno.cz
Sources
1. Vencovský J. Idiopatické zánětlivé myopatie. Vnitř Lék 2018; 64(2): 155–163.
2. Vencovský J. Idiopatické zánětlivé myopatie – některé novější aspekty. Neurol Praxi 2020; 21(6): 477–484.
3. Carstens PO, Schmidt J. Diagnosis, pathogenesis and treatment of myositis: recent advances. Clin Exp Immunol 2014; 175(3): 349–358. doi: 10.1111/ cei.12194.
4. Rogers A, Chung L,Shufeng L et al. The cutaneous and systemic findings associated with nuclear matrix protein-2 antibodies in adult dermatomyositis patients. Arthritis Care Res 2017; 69(12): 1909–1914. doi: 10.1002/ acr.23210.
5. Inoue M, Tanboon J, Hirakawa S et al. Association of dermatomyositis sine dermatitis with anti-nuclear matrix protein 2 autoantibodies – NXP2. JAMA Neurol 2020; 77(7): 872–877. doi: 10.1001/ jamaneurol.2020. 0673.
6. Butt Z, Patel L, Das MK et al. NXP-2 positive dermatomyositis: a unique clinical presentation. Case Rep Rheumatol 2017; 2017: 4817275. doi: 10.1155/ 2017/ 4817275.
7. Schmidt JJ. Current classification and management of inflammatory myopathies. Neuromuscul Dis 2018; 5(2): 109–129. doi: 10.3233/ JND-180308.
8. Allenbach Y, Benveniste O. Diagnostic Utility of auto antibodies in inflamatory muscle disease. J Neuromuscul Dis 2015; 2(1): 13–25.
9. Yan T, Zhang X, Yang H et al. Association of anti-NXP2 antibody with clinical characteristics and outcomes in adult dermatomyositis: results from clinical applications on a myositis.specific antibody. Clin Rheumatol 2021; 40(9): 3695–3702. doi: 10.1007/ s10067-021-05 667.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2022 Issue 2
Most read in this issue
- Cílená chirurgická léčba obstrukční spánkové apnoe
- Poruchy rovnováhy u osob s roztroušenou sklerózou a možnosti rehabilitační terapie – aktuální poznatky kontrolovaných klinických studií
- Úspěšná konzervativní terapie radikulopatie v terénu objemné hernie disku s nestabilitou u low back pain syndromu
- Muzikoterapia pri poruchách hlasu a reči u pacientov s Parkinsonovou chorobou