
Nálezy na magnetické rezonanci u neuromyelitis optica a onemocnění jejího širšího spektra
Magnetic resonance imaging in neuromyelitis optica spectrum disorders
Neuromyelitis optica is an autoimmune disease of unknown etiology characterized by primary involvement of the optic nerves and spinal cord. Unlike in MS, astrocytes are primarily affected. Lesions on MRI are mainly located in areas where aquaporin-4 is overexpressed. Lesions can be found in diencephalon periependymally around the lateral ventricles or fourth ventricle and in the corpus callosum. Pyramidal pathway involvement may also be seen but it is not associated with increased aquaporin-4 expression. Intramedullary involvement is most frequent in the form of longitudinal extensive transverse myelitis, where the lesion extends over three or more vertebral bodies. These lesions primarily affect the gray matter, often extend into periphery and occupy more than 50% of the spinal cord area in transverse cuts. Optic nerve involvement is typically bilateral in the dorsal part, also affecting the chiasma.
Authors:
M. Vaněčková
Authors‘ workplace:
Oddělení MR, Radiodiagnostická klinika 1. LF UK a VFN v Praze
Published in:
Cesk Slov Neurol N 2020; 83/116(supplementum 1): 20-30
doi:
https://doi.org/10.14735/amcsnn2020S20
Overview
Neuromyelitis optica je autoimunitní onemocnění neznámé etiologie, které je charakterizováno primárním postižením optických nervů a míchy. Na rozdíl od RS jsou zde primárně postiženy astrocyty. Ložiska na MR jsou především v oblastech, kde je zvýšeně exprimován akvaporin-4. Jedná se o ložiskové postižení v oblasti diencefalon, periependymálně v okolí postranních komor nebo IV. komory a v oblasti corpus callosum. Může být také patrné postižení pyramidové dráhy, které však není spjato se zvýšenou expresí akvaporinu-4. Intramedulární postižení je nejčastěji ve formě longitudinální extenzivní transverzální myelitidy, kdy ložisko svojí délkou dosahuje výšky třech nebo více obratlových těl, ložisko primárně postihuje šedou hmotu, často rozsáhle zasahuje i periferně a na transverzálních řezech zaujímá více než 50 % plochy míchy. Postižení zrakových nervů je typicky bilaterální v dorzální části postihující i chiasma.
Úvod
Neuromyelitis optica (NMO) nebo také Devicova nemoc je autoimunitní onemocnění neznámé etiologie, které je charakterizováno primárním postižením optických nervů a míchy a většinou rychlým a agresivním průběhem [1]. Na rozdíl od RS jsou zde primárně postiženy astrocyty (u RS oligodendrocyty). NMO je vzácné onemocnění a po zlepšení diagnostických možností, především testováním protilátek proti akvaporinu-4 (AQP4-IgG), se zjistilo, že onemocnění bylo v Evropě poddiagnostikováno. Pacienti byli často mylně vedeni pod diagnózou RS. Odhaduje se, že NMO v Evropě tvoří přibližně 1,5 % ze získaných demyelinizačních onemocnění. NMO může mít monofázický nebo recidivující průběh onemocnění. Ženy jsou stejně jako u RS postiženy častěji než muži, a to ve vyšším poměru než u RS. Odlišností oproti RS je, že NMO nemá typickou sekundárně progresivní fázi. Pacient je ohrožen zejména těžkými relapsy, často s rezidui, po kterých může být několikaleté období remise. Na rozdíl od RS MR není schopna predikovat průběh onemocnění [2].
Jednou z prvních zmínek o tomto onemocnění byla práce Allbutta z roku 1870, která ukázala na spojitost mezi myelitidou a optickou neuritidou. Následně Devic roku 1894 zavedl termín akutní optická neuromyelitida [3]. Důležitým mezníkem v diagnostice NMO byl rok 2004, kdy byly objeveny tzv. NMO-IgG, které jsou specifické pro toto onemocnění. Nedlouho poté byla stanovena cílová struktura, proti které jsou protilátky namířeny, a to AQP4. Pro NMO je typická přítomnost AQP4-IgG v séru. Jejich senzitivita je přibližně 80 % a specificita až 100 %. AQP4 je ubikvitní vodní kanál lokalizovaný v různých tělesných strukturách (CNS, ledviny). AQP4 je exprimován zejména astrocyty v oblasti perivaskulární, subependymální a glia limitans [4,5]. Dobře známá jsou kritéria z roku 2006, která definovali Wingerchuk et al a u kterých byla zahrnuta pozitivita těchto protilátek. Pro jisté onemocnění bylo nutné splnit dvě absolutní kritéria (optická neuritida, akutní myelitida) a z podpůrných kritérií alespoň dvě ze tří: 1. intramedulární ložisko, jehož délka dosahuje alespoň tří segmentů; 2. normální nález na MR mozku nebo nález nesplňující Barkhofova kritéria pro RS v rámci revidovaných McDonaldových kritérií; 3. séropozitivita NMO-IgG (po objevení cílového antigenu se začalo používat označení AQP4-IgG – pozn. autora) [3]. V roce 2015 Wingerchuck et al (Mezinárodní panel odborníků pro NMO diagnózu) publikovali nová diagnostická kritéria pro NMO a onemocnění jejího širšího spektra (neuromyelitis optica spectrum disorders; NMOSD), tato kritéria již akceptují klinickou manifestaci při postižení lokalit mimo optické nervy a míchu (např. postižení mozkového kmene a další) (tab. 1). Nová kritéria rozdělují NMOSD s pozitivitou protilátek proti AQP4-IgG a bez pozitivity [4]. Pro správnou diagnostiku, především pro odlišení od RS, je nutné znát typický nález na MR u NMO (tab. 2) [5]. Odlišení těchto dvou onemocnění má zcela zásadní význam pro správný terapeutický postup, který se u těchto dvou jednotek liší. Asi u 5–40 % pacientů s AQP4-IgGnegNMOSD nacházíme v séru jiné protilátky namířené proti myelinovému oligodendrocytárnímu glykoproteinu (MOG-Ig).
Magnetická rezonance mozku, zrakových nervů a míchy hraje jednu z hlavních rolí v diagnostickém procesu NMOSD. V mezinárodním konsenzu diagnostických kritérií pro NMOSD z roku 2015 jsou uvedena konkrétní rezonanční kritéria pro daný klinický obraz. Provedení vyšetření MR je podstatné z důvodu vyloučení jiné diagnózy či koincidenci různých onemocnění. Pokud klinické obtíže pacienta odpovídají tzv. jádrovým klinickým charakteristikám typickým pro NMOSD (např. optická neuritida, akutní myelitida, syndrom area postrema a další), ale nenacházíme v séru AQP4-IgG, pak je vyžadováno naplnění požadavků MR pro stanovení diagnózy AQP4-IgGnegNMOSD [4]. Konkrétní požadavky MR jsou pak uvedeny v tab. 3.
Nález na MR
Ložiska na MR jsou u pacientů s NMOSD především v oblastech, kde je zvýšeně exprimován AQP4, ale patologická ložiska mohou být i v oblastech, které s ní nejsou spjaty. Ložiska v oblasti diencefalon, periependymálně v okolí postranních komor nebo IV. komory, v oblasti corpus callosum (CC) nebo v oblasti kortikospinálního traktu podporují tuto diagnózu (tab. 2) [4,5].
Incidence patologických ložisek v mozkovém parenchymu je reportována od 50–85 % s použitím NMO diagnostických kritérií z roku 2006. Při užití kritérií z roku 2015 pro séropozitivní AQP4-IgGpozNMOSD je to 51–89 % [6]. Na začátku onemocnění jsou popisována ložiska v oblasti mozkového parenchymu u 43–70 % pacientů s NMOSD [7]. Výskyt těchto ložisek dále stoupá s délkou trvání nemoci [6].
Ložiska v oblasti diencefalon v okolí III. komory a aqueductu
Léze v oblasti diencefalon zahrnují periependymální oblast mezimozku přilehlou k aqueductu a talamus a hypotalamus přilehlý k III. komoře (obr. 1 a, b). Tyto oblasti korespondují s vysokou expresí AQP4. Tento vzorec postižení je typický pro NMOSD, ale není typický pro RS. Symptomatická narkolepsie nebo akutní diencefalický syndrom s ložiskovým postižením na MR je jednou z jádrových klinických charakteristik u revidovaných kritérií z roku 2015 [4,6,8].
FIG.1 AQP4-IgGposNMOSD patient, lesions near the aqueduct.
(a) T2 weighted imaging – transversal cut, hypersignal lesion near the aqueduct (arrow);
(b) FLAIR – diff use hypersignal near the aqueduct (arrow).
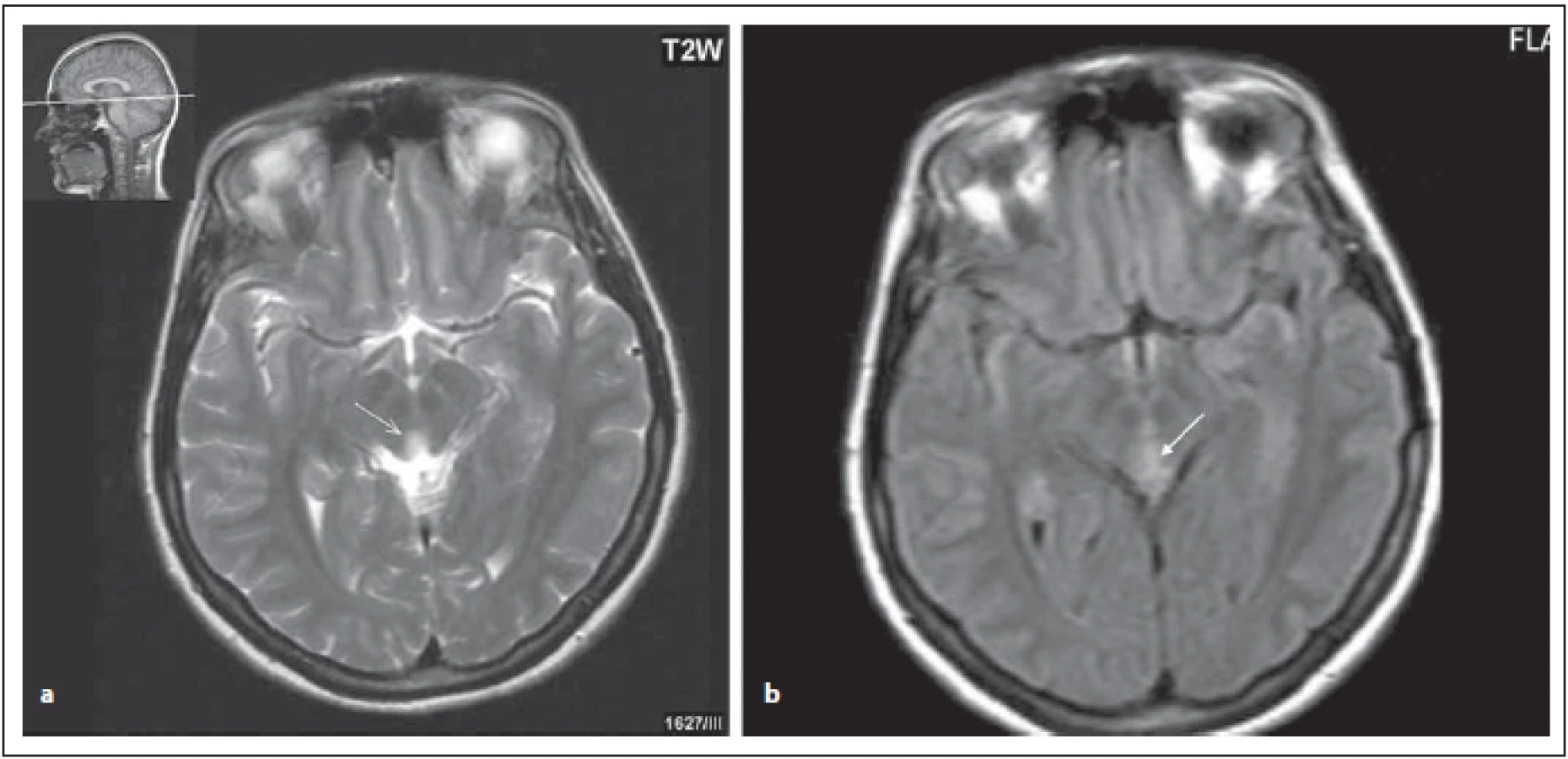
FLAIR – fl uid attenuated inversion recovery; NMOSD – neuromyelitis optica spectrum disorder
Ložiska v dorzální části kmene v okolí IV. komory
Pro NMOSD jsou typická periependymální ložiska v zadní části kmene a mozečku zasahující k IV. komoře (obr. 2 a, b). Jedna z oblastí, která je vysoce specifická pro toto onemocnění, je area postrema a nucleus tractus solitarius (ložiska se vyskytují u 7–46 % pacientů s NMOSD), čemuž klinicky odpovídá syndrom area postrema charakterizovaný protrahovaným singultem nebo nauzeou se zvracením, které patří mezi jádrové charakteristiky kritérií revidovaných v roce 2015 (obr. 3 a, b) [4,6]. Ložiska v prodloužené míše bývají drobná, lineární, bilaterální a mohou přecházet na krční míchu. Ložiska v této oblasti mohou asociovat s prvním klinickým příznakem nebo s akutní exacerbací onemocnění. Mimo výše popsané postižení dorzální části oblongáty bývá přítomno i postižení dorzálního pontu a mozečku (obr. 2 a, b). U séronegativních pacientů s kmenovým syndromem má pro diagnózu NMOSD velký význam zobrazení periependymálních ložisek v oblasti mozkového kmene [7].
Fig. 2. AQP4-IgGposNMOSD patient, T2-weighted image, diff use hypersignal in cerebellar peduncles (arrows), transversal cut (a),
adjacent to the fourth ventricle on the left side (for detail, see fi gure b (arrow)), discrete hypersignal lesions in ventral pons.
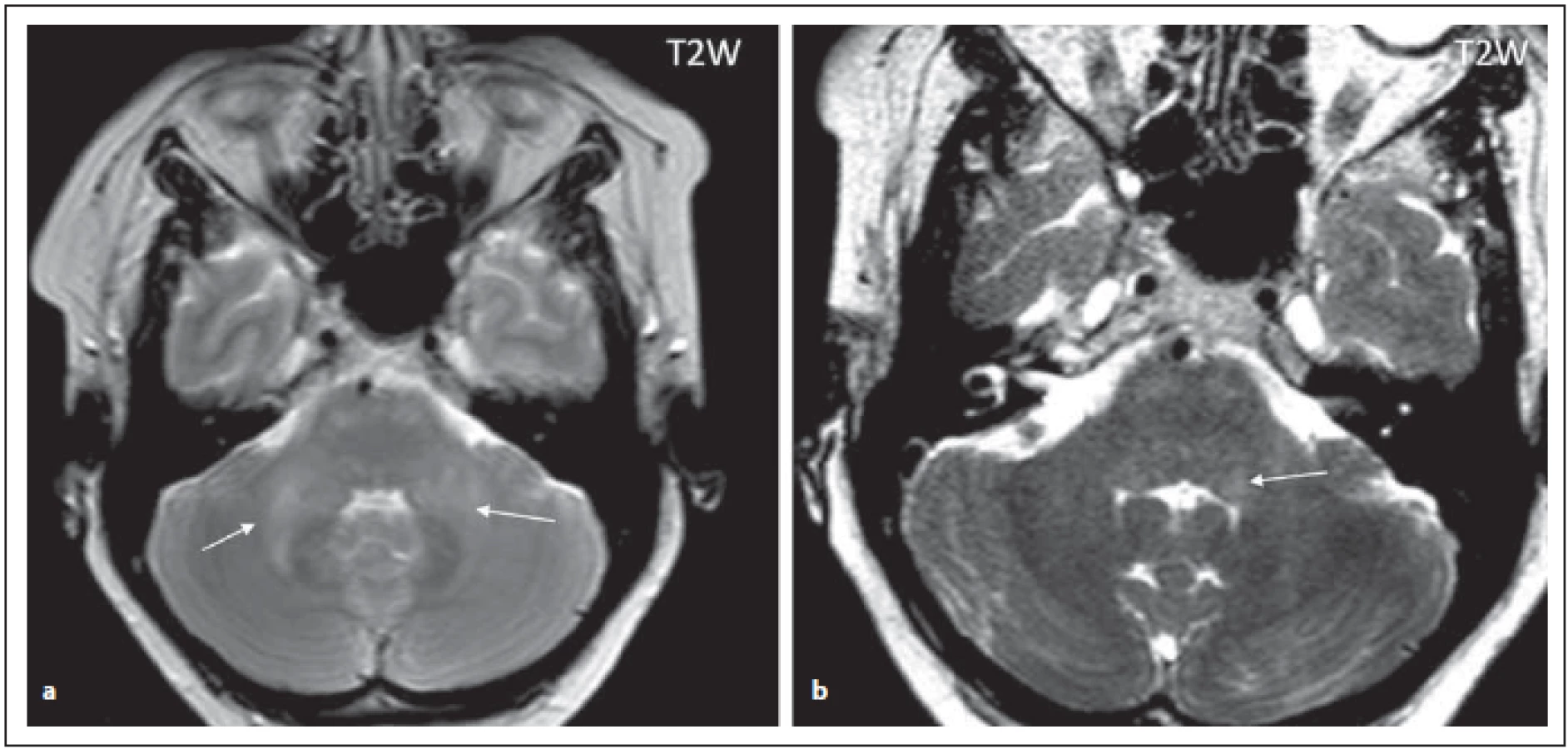
NMOSD – neuromyelitis optica spectrum disorder
Fig. 3. MOG-IgGposNMOSD patient, T2-weighted image, sagittal cut, hypersignal lesion in dorsal oblongata (area postrema, tractus
solitarius) (arrow); high signal in middle cerebellar peduncle, in pons and mesencephalon (a). Lesions in dorsal oblongata extend to
nucleus tractus solitari bilaterally (b). There is a lesion in corpus callosum with a diff erent shape than in MS patients (dashed arrow) (a).
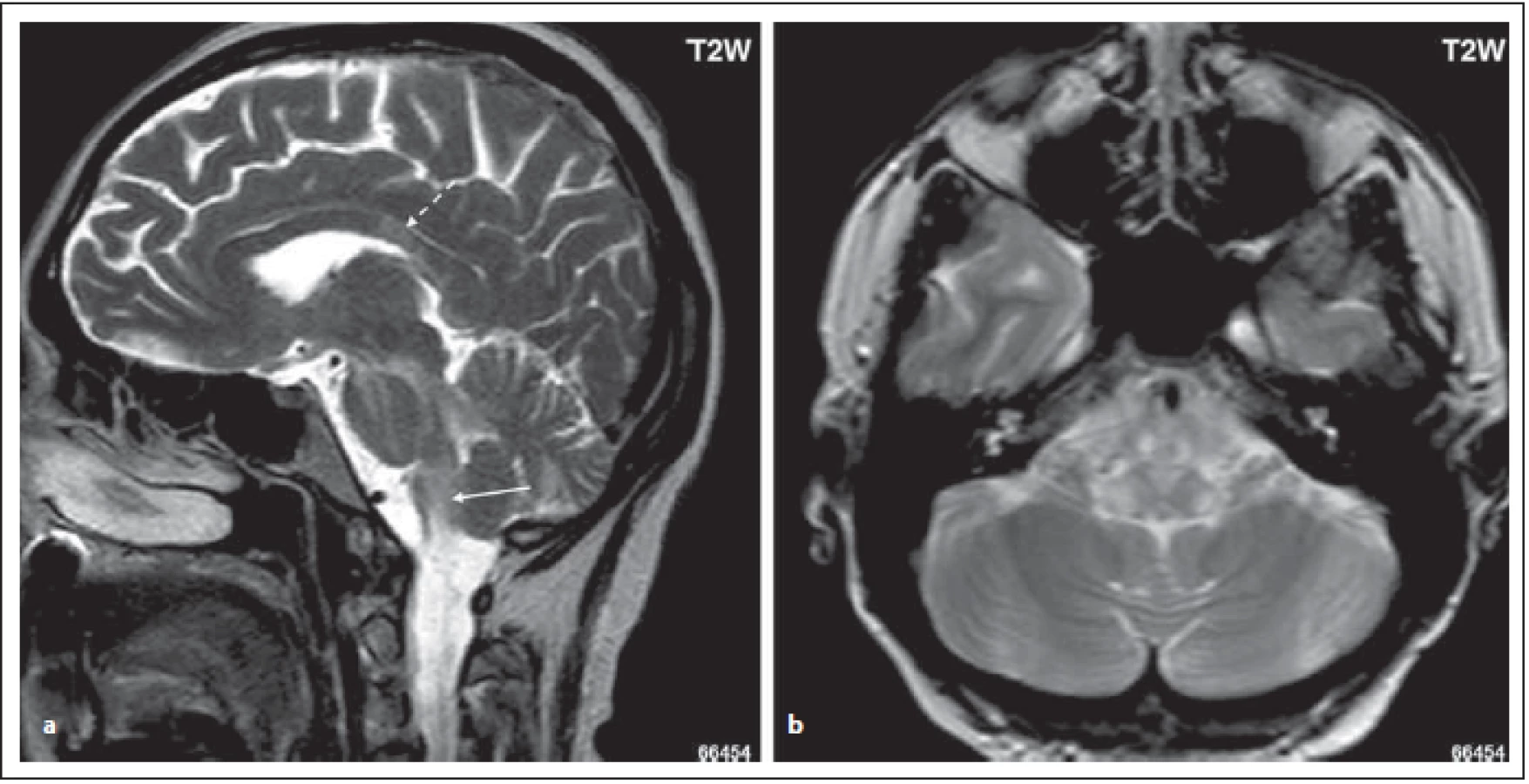
NMOSD – neuromyelitis optica spectrum disorder
Ložiska v oblasti bílé hmoty mozkových laloků
Postižení v bílé hmotě může být značně rozsáhlé, ložiska jsou často větší než 3 cm, nebývá přítomen mass efekt. Ložiska mohou být uni- nebo bilaterální, zahrnující subkortikální nebo hlubokou bílou hmotu (obr. 4 a–c). Tvar může být vřetenovitý nebo paprsčitý. Zvyšuje signál na ADC mapě, což je korelátem vazogenního edému s akutní demyelinizací. Tato rozsáhlá ložiska se vyskytují častěji u AQP4-IgGpoz než u AQP4-IgGneg pacientů. Výskyt se liší dle jednotlivých prací, Kim et al tyto léze popsali u 29 % pacientů, v práci Wang et al jen ve 14 % [5,6]. V chronických fázích se mohou zmenšovat a objevit se cystické změny [5] (obr. 4 b, c).
Fig. 4. AQP4-IgGposNMOSD patient, FLAIR, extensive hypersignal lesion in the right frontal lobe without edema, no mass eff ect (a), partial
cavitation of the lesion at monthly examination (b), signifi cant reduction of the lesion at 2-year examination (c).
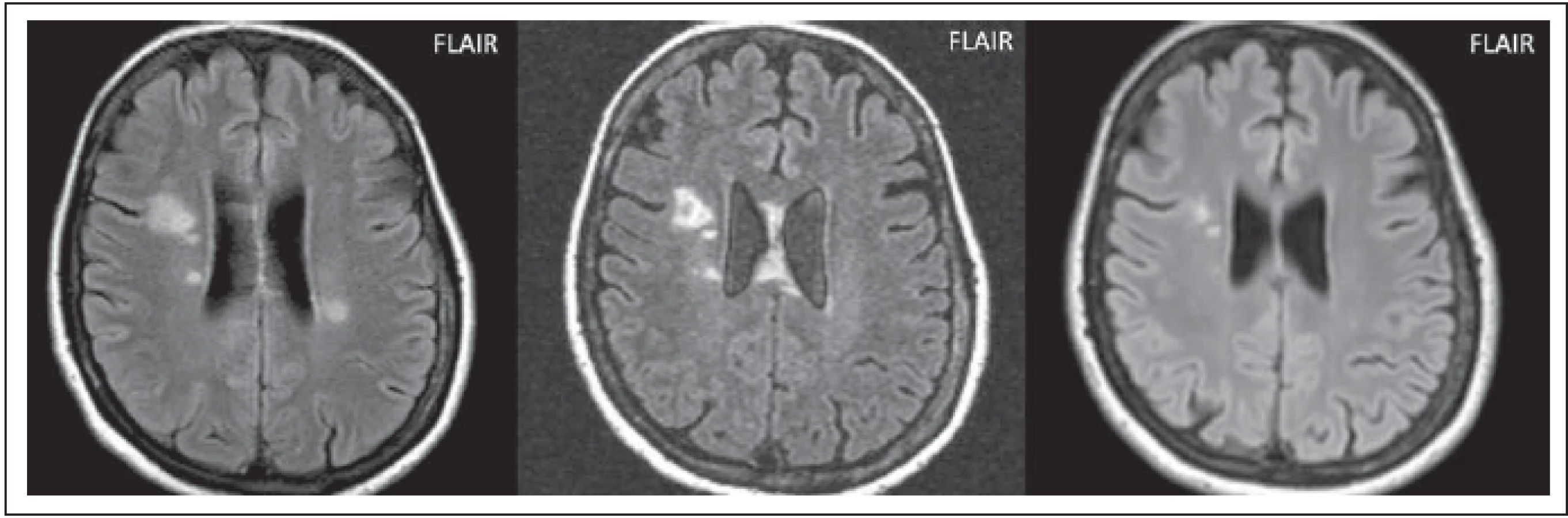
FLAIR – fl uid attenuated inversion recovery; NMOSD – neuromyelitis optica spectrum disorder
Ložiska v okolí postranních komor
Ložiska v okolí postranních komor jsou u NMOSD shledávána méně často než v blízkosti III. a IV. komory. Ložiska jsou uložená paralelně s průběhem komory na rozdíl od kolmého uspořádání u RS (obr. 5 a, b). U NMOSD bývají u 12–40 % pacientů ložiska v oblasti CC, která bývají často edematózní, heterogenní na rozdíl od ovoidních ložisek u RS (obr. 6 a, b). Někdy je postižení CC připodobňováno k mramorování, někdy je postižení rozsáhlejší, je patrné ztluštění splenia corporis callosi – vzorec postižení jako mostní klenba. V chronické fázi mají CC ložiska tendenci se zmenšovat, mohou vyvolat cystické změny, nebo mohou i vymizet, často později dochází k atrofii corporis callosi (obr. 6 c) [5,6].
Fig. 5. AQP4-IgGposNMOSD patient, FLAIR, periventricular hypersignal in the right temporal horn of lateral ventricle (a), high signal
periependymally in the left lateral ventricle (b), nonspecifi c lesion in centrum semiovale.
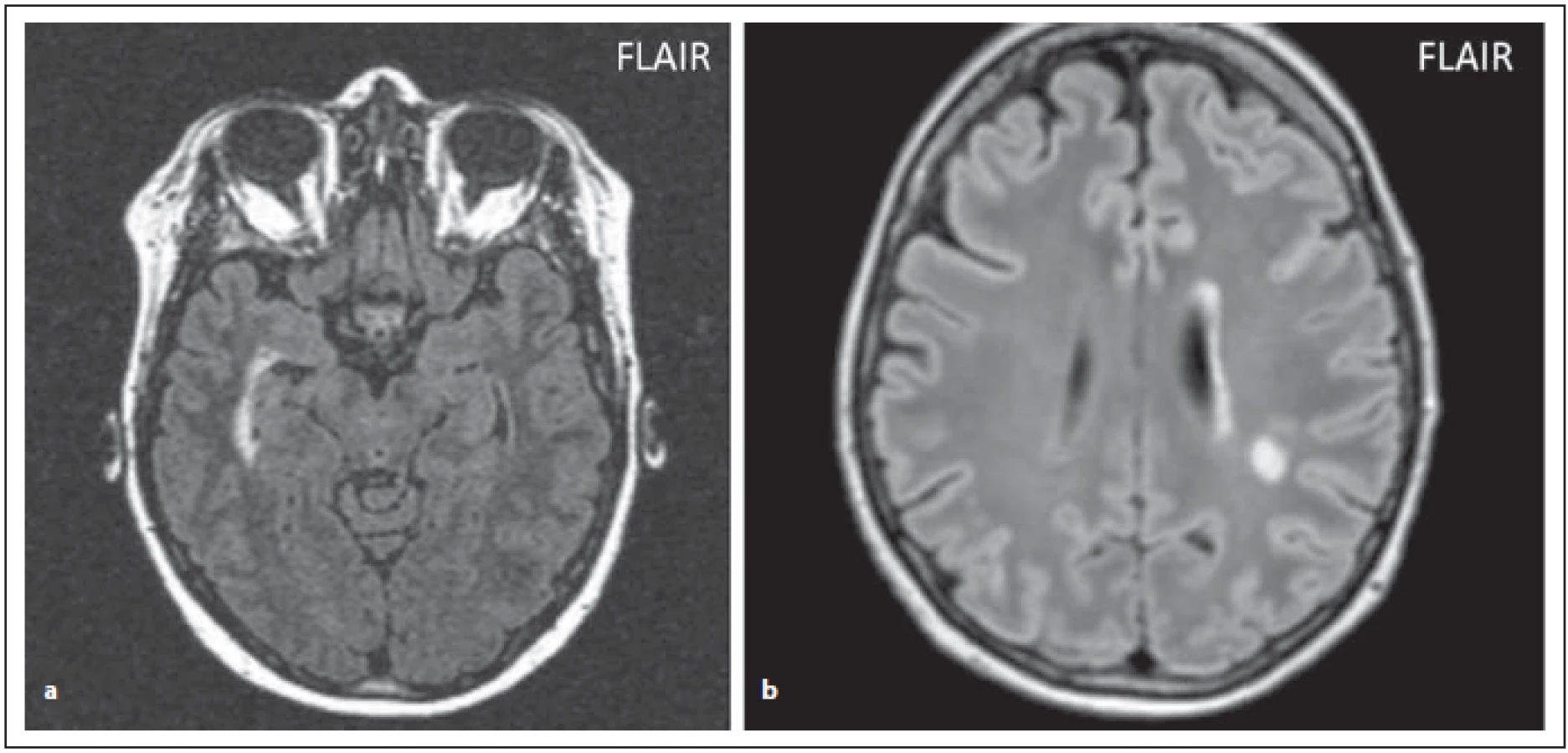
FLAIR – fl uid attenuated inversion recovery; NMOSD – neuromyelitis optica spectrum disorder
Fig. 6. AQP4-IgGposNMOSD patient, T2-weighted image, sagittal cut, multiple (and more extensive in some places) lesions
in corpus callosum (a), FLAIR, extensive lesion in the splenium (b). Signifi cant atrophy of the corporis callosi is observed at 2-year
follow-up (c).
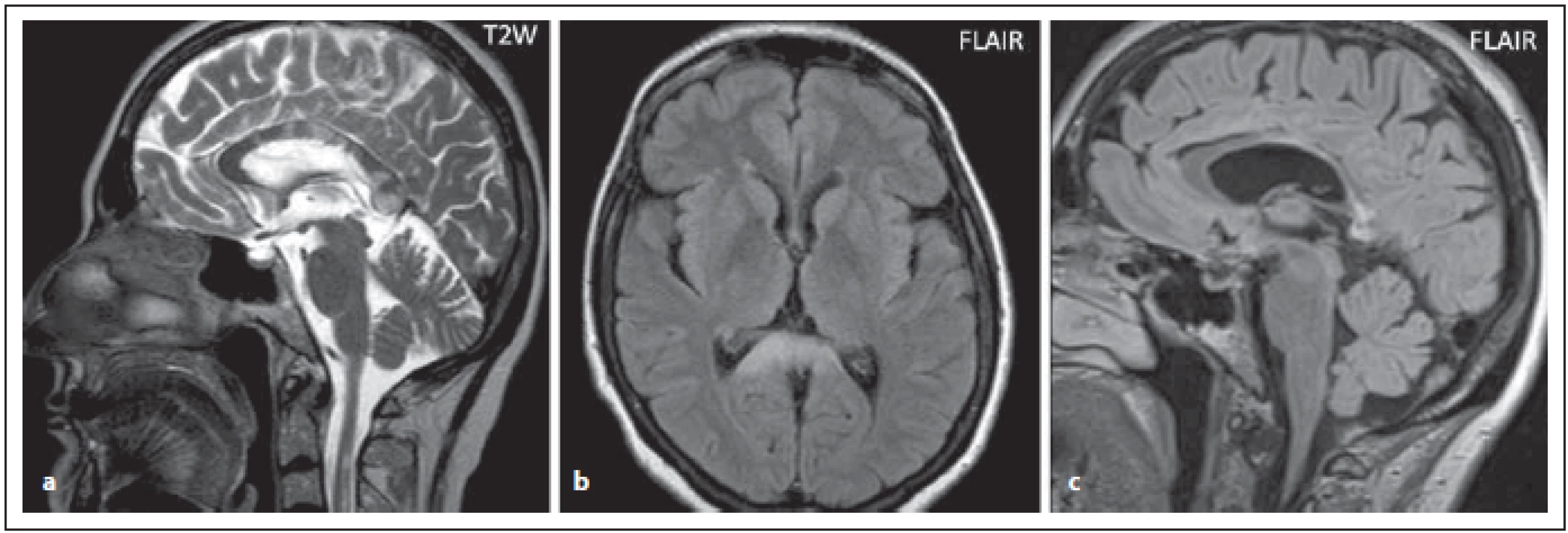
FLAIR – fl uid attenuated inversion recovery; NMOSD – neuromyelitis optica spectrum disorder
Ložiskové postižení kortikospinálního traktu
U určitých podskupin pacientů s NMOSD může být postižen kortikospinální trakt, a to buď uni- nebo bilaterálně. Ložiska zasahují do hluboké bílé hmoty, je patrný zvýšený signál v oblasti zadního raménka kapsuly interny a zasahují až do cerebrálních pedunkulů. Tato lokalizace není spojená se zvýšenou expresí AQP4 a do současnosti nebylo objasněno, proč je pyramidová dráha postižena (obr. 7 a–c) [5–8].
Fig. 7. MOG-IgGposNMOSD patient, the same patient as in Fig. 3. High signal is evident in T2-weighted image bilaterally in the pyramidal
tract – in the dorsal limb (a, b), in cerebral peduncles (c).
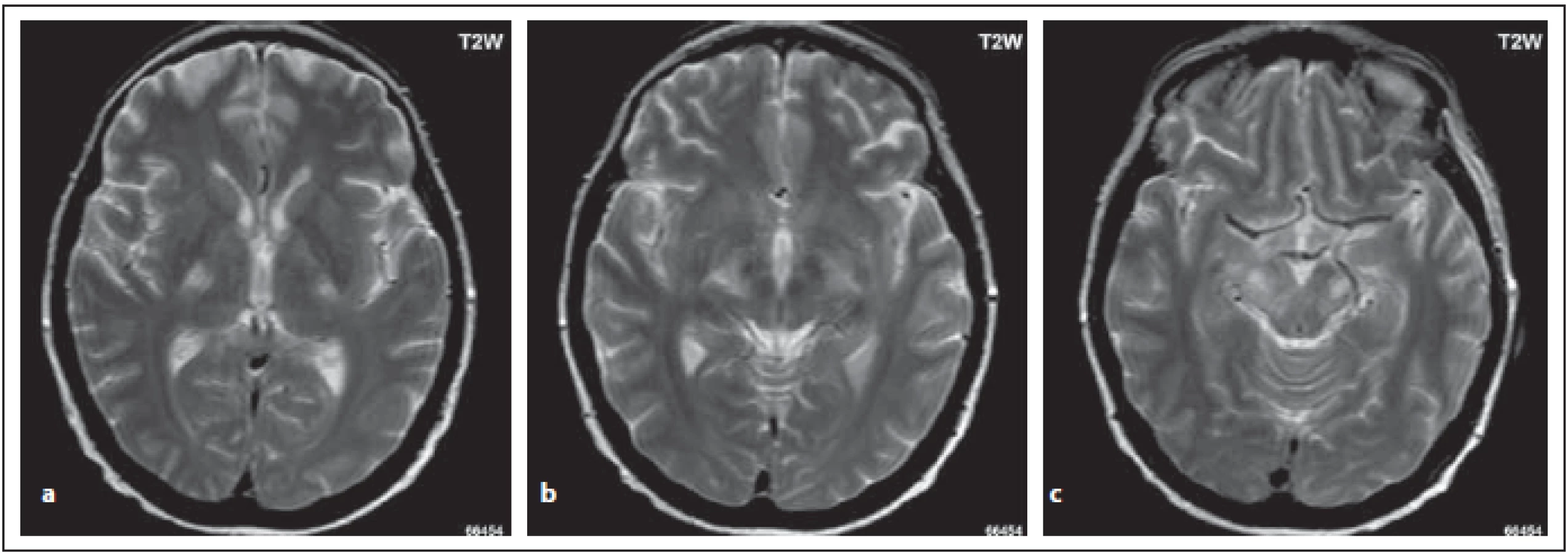
NMOSD – neuromyelitis optica spectrum disorder
Nespecifické ložiskové postižení bílé hmoty supratentoriálně
Drobnoložiskové postižení bílé hmoty je u NMOSD relativně časté a je zcela nespecifické. Jedná se o vícečetná drobná hypersignální ložiska v T2 váženém obraze a FLAIR v hluboké bílé hmotě nebo subkortikálně. Tato ložiska bývají u NMOSD asymptomatická [5,6].
Enhancement po podání kontrastní látky
Po podání kontrastní látky je variabilní přítomnost enhancementu (vyskytuje se v 9–36 %) [6,9]. Na rozdíl od RS je diskrétní, hůře ohraničený, připodobňuje se k obláčkům („cloud-like“), dále může být patrný enhancement ependymu komor, který se připodobňuje k obtažení tužkou. Může být přítomen také nodulární enhancement, prstenčitý, leptomeningeální a byl popsán i perivaskulární (obr. 8 a–c) [8,9].
Fig. 8. AQP4-IgGposNMOSD patient, T2-weighted image, sagittal cut, intramedullary hypersignal lesion is evident, which slightly expands
the spinal cord, its length exceeds the height of three vertebral bodies – at the level from C6 to the middle Th (a) – so-called
LETM, slightly hyposignal, sometimes more pronounced in T1-weighted image (b). The lesion has central localization. After contrast
agent administration, irregular (similar to small clouds) enhancement, mostly marginal postcontrast enhancement (c).
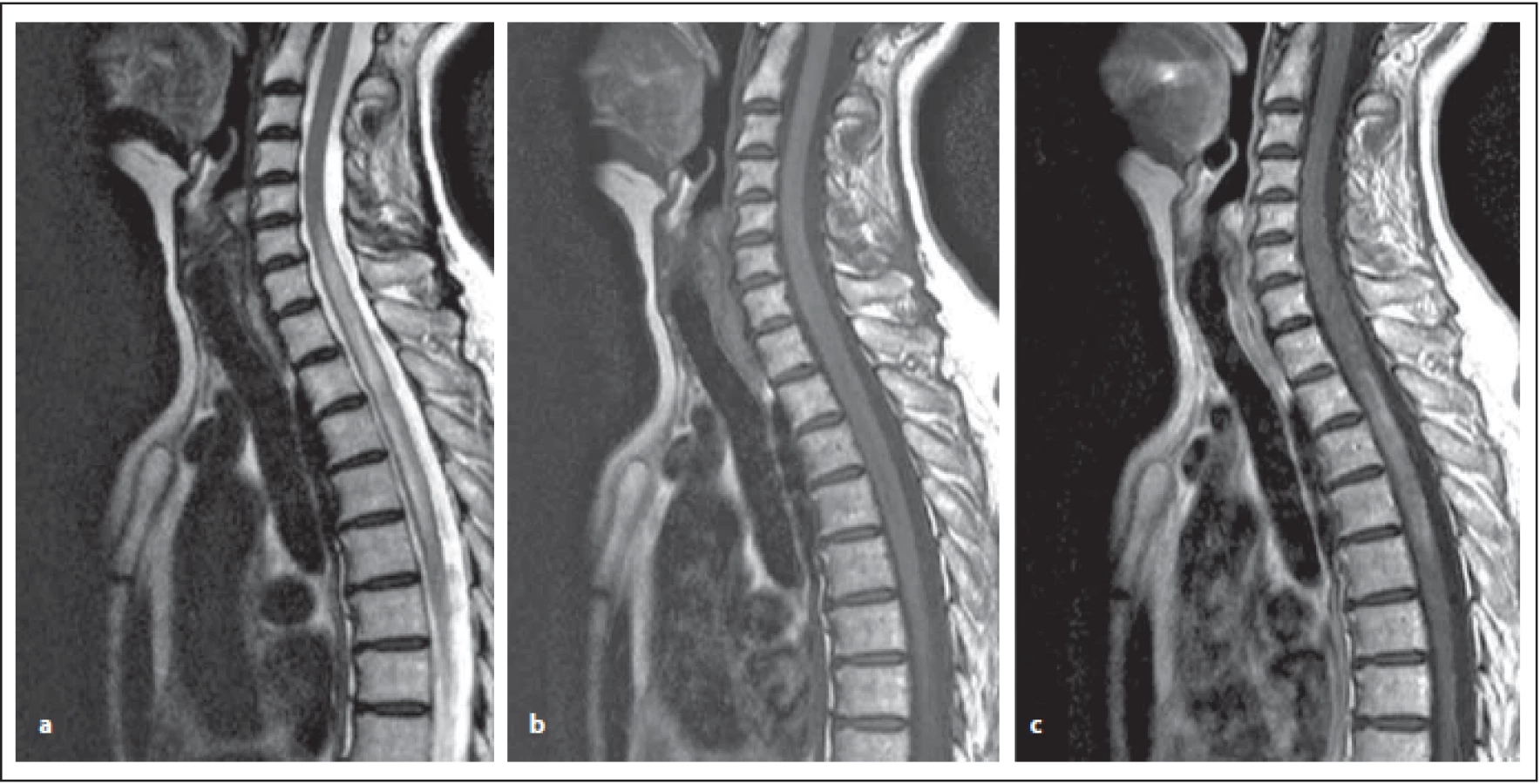
NMOSD – neuromyelitis optica spectrum disorder
Intramedulární ložiska
Ložiska v oblasti míchy jsou rozsáhlejší než u RS, typicky svojí délkou dosahují nebo přesahují výšku 3 obratlových těl, mluvíme o tzv. longitudinálně extenzivní transverzální myelitidě (LETM). Ložiska jsou typicky uložena centrálně v šedé hmotě, podél centrálního kanálku. Tato predilekční area koresponduje s výraznější expresí AQP4 v této oblasti [10,11].
Na MR jsou ložiska patrná centrálně nebo jsou rozsáhlejší, potom zasahují i periferně na transverzálních řezech a zahrnují více než 50 % plochy míchy na transverzálních řezech [12]. Ložiska jsou hypersignální v T2 váženém obraze, v akutním stadiu je přítomen edém, v T1 vážených obrazech jsou léze hypointenzní a mohou se zvýrazňovat po podání kontrastní látky. Enhancement bývá spíše nepravidelný (připodobňovaný k obláčkům), nebo bývá kombinací nepravidelného s prstenčitým (obr. 8 a–c). Výrazně hypersignální ložisko v T2 váženém obraze, respektive s přítomností výrazně hypersignálních teček, korespondující s hyposignálními ložisky v T1 váženém obraze, je typické pro NMOSD – ne pro RS. Nejčastěji jsou léze v oblasti krční a horní hrudní míchy. Typické rozsáhlé intramedulární ložisko se může v průběhu onemocnění měnit, v období remise či po léčbě vysokými dávkami kortikoidů se může zobrazovat v podobě vícečetných drobných ložisek, která již nesplňují délkou kritéria pro LETM (obr. 9 a, b) [6,12]. Také u pacientů, kteří již jsou na chronické imunosupresivní léčbě pro jiné autoimunitní onemocnění nebo předcházející optické neuritidy, můžeme vidět ložiska kratší [10]. Po opakovaných myelitidách je často patrná atrofie míchy, která koreluje s neurologickým postižením. Možnost splnění kritérií (bez přítomnosti LETM) při přítomnosti lokální atrofie míchy v rozsahu alespoň přes 3 segmenty reflektují revidovaná kritéria z roku 2015 [4].
Fig. 9. AQP4-IgGposNMOSD patient,
T2-weighted image, sagittal cut,
atrophy in thoracic spinal cord and multiple
hypersignal lesions in thoracic spinal
cord are evident (a), typical central localization
of the lesion in transversal cuts
is observed (b).
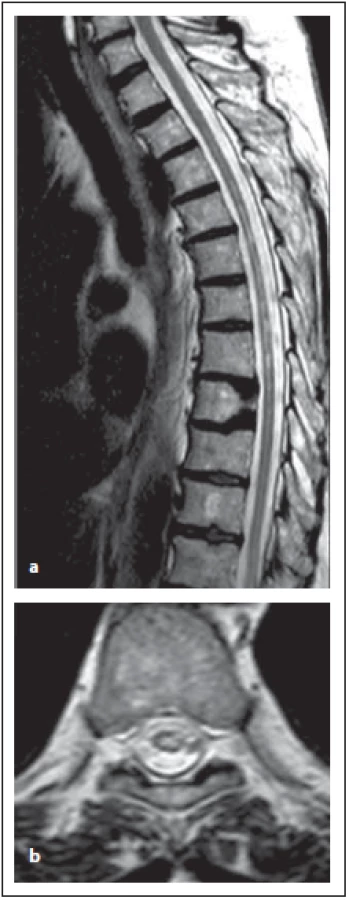
NMOSD – neuromyelitis optica spectrum
disorder
Někdy jsou ložiska již primárně drobnější, hovoří se o krátké transverzální myelitidě (short transverse myelitis; STM), která je definována jako ložisko kratší, než je výška 3 obratlových těl. Tato ložiska nejsou u NMOSD výjimečná, v iniciálních stadiích se mohou vyskytovat až ve 14,5 % [7,13,14]. Tato ložiska opět preferenčně postihují centrální šedou hmotu a postihují více než 50 % arey na transverzálních řezech, což pomáhá v odlišení od ložisek u RS [6].
Postižení optického nervu
Optická neuritida je v MR obraze charakterizována rozšířením nervus opticus, zvýšeným signálem v T2 váženém obraze a enhancementem po podání kontrastní látky. Typické je bilaterální postižení, ložiska jsou dlouhá (zasahují více než 50 % délky) [6,8,15]. Pro NMOSD je na rozdíl od RS typické postižení chiasmatu. V chronickém stadiu je patrná atrofie optického nervu s variabilně zvýšeným signálem v T2 váženém obraze [10,15].
„MOG antibody disease“ (MOG-AD, onemocnění asociovaná s MOG-IgG) nebo též MOG encefalomyelitis
MOG-AD se klinicky může manifestovat jako NMOSD, akutní diseminovaná encefalomyelitida (ADEM), ADEM následovaný ON (ADEM-ON) nebo multifázická diseminovaná encefalomyelitida (MDEM). Toto spektrum onemocnění může postihnout pacienty všech věkových dekád, medián objevení se onemocnění je třetí dekáda s mírnou převahou u žen (ženy/muži 1,1: 1) [16,17]. U více než 50 % dětských pacientů s ADEM nacházíme v séru MOG-IgG a predikují non-RS průběh onemocnění. Vzácněji může být onemocnění asociováno s demyelinizační NMDA encefalitidou (N-methyl-d-aspartate receptor encephalitis), postiinfekčně po proběhlé herpes virové infekci, po borelióze, mononukleóze a velmi zřídka s typickou relaps remitentní RS.
Optická neuritida je nejčastěji se objevující klinické postižení u MOG-AD a to v 54–61 %, následované myelitidou [8]. Jsou určité odlišnosti týkající se MR nálezu u pacientů s pozitivitou MOG-IgG nebo AQP4-IgG. Zánět optického nervu predominantně postihuje přední segment, zatímco chiasma a retrochiasmatický úsek jsou relativně ušetřeny. U AQP4-IgG pacientů je predominantně postižená oblast chiasmatu [15]. Optický nerv bývá edematózně rozšířen, edém zrakového disku bývá zvlněný. Až u poloviny pacientů se může objevit zánět a enhancement pochvy zrakového nervu. Oboustranné postižení zrakových nervů bývá u čtvrtiny pacientů. Výše popsané zánětlivé postižení bývá patrné v akutní fázi, po jejím odeznění u většiny pacientů přetrvává zvýšený signál v T2 váženém obraze a objevuje se atrofie optického nervu [17]. Postižení míchy má charakteristické dva vzorce postižení: 1. ve formě LETM; 2. ve formě kratších lézí (méně než přes dvě obratlová těla) [18] (obr. 10 a, b). Postižení míšního konu je typické pro myelitidy asociované s MOG-IgG, ale může postihovat míchu i v celém jejím rozsahu. Enhancement bývá typicky mírný, nepravidelný, mícha je často rozšířená. Může být i nodulární enhancement či enahncement mening. Ložisko se obvykle nachází v blízkosti centrálního kanálku, je výrazně hypersignální v T2 váženém obraze, někdy popisováno jako „pseudodilatace centrálního kanálku“, což je typické pro MOG-IgG, ale ne pro RS či AQP4-IgGpozNMOSD. Přítomný může být enhancement tohoto ložiska.
Fig. 10. MOG-IgGposNMOSD patient,
sagittal T2-weighted image, transversal
cut, intramedullary lesion in Th11–12 segment
(a), with typical central localization
in the transversal cut (b).
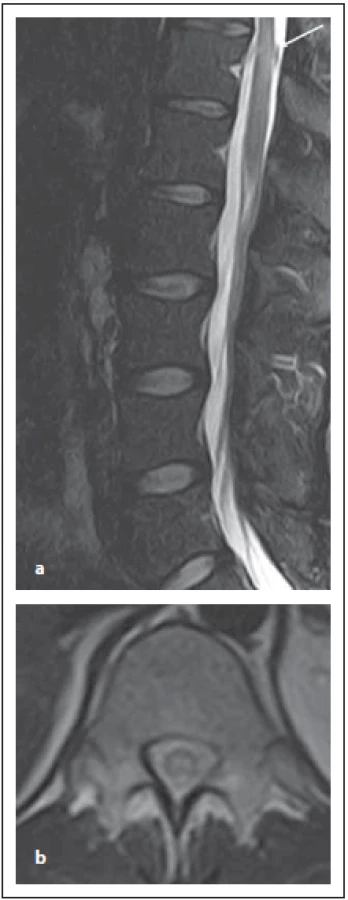
MOG-IgG – myelin oligodendrocyte glycoprotein
antibody; NMOSD – neuromyelitis
optica spectrum disorder
Negativní nález na MR mozku je přibližně u dvou třetin pacientů nebo bývají přítomna nespecifická ložiska v bílé hmotě supratentoriálně, spíše subkortikálně. Tato nespecifická ložiska bývají asymptomatická [8,17]. Méně často mohou být přítomna atypická ložiska infratentoriálně: v mezencefalon, pontu, mozečkových pedunkulech [15]. Někdy může být obraz napodobující ADEM, kde jsou patrné difuzní změny kortiko/subkortikálně, v bílé hmotě, hluboké šedé hmotě. V diferenciální diagnostice s RS pomáhá tvar enhancementu, protože ten bývá nepravidelný, připodobňovaný k obláčkům – na rozdíl od prstenčitého u RS. Dále pomáhá diskriminovat MOG-AD leptomeningeiální enhancement a ložiska v talamech [19].
V práci Ikeda et al bylo u dětské populace popsáno ložiskové postižení v subkortikální bílé hmotě. Ložiska byla jak ostře ohraničená tak i neostře ohraničená, popisovaná spíše jako difuzní změna signálu. Jiná distribuce ložisek nebyla zjištěna, ale jednalo se jen o malou skupinu 4 pacientů [20]. Další práce udávaly vzorec MR postižení jako „ADEM like“, kde byla ložiska i v hluboké bílé hmotě, talamu a bazálních gangliích [21]. U MOG-AD nebývají přítomna ložiska v oblasti CC a ložiska nebývají periventrikulárně, což pomáhá v odlišení od RS. Byla popsána distribuce lézí v závislosti na věku dětských pacientů – mladší 9 let se prezentují jako ADEM, u starších se většinou rozvíjí optická neuritida [20].
Rozdíly v MR nálezu u RS a NMOSD
Na rozdíl od RS nejsou u NMOSD typicky přítomna kortikální ložiska. Z tohoto důvodu by měl diagnostický protokol obsahovat sekvence, které umožňují jejich detekci, např. double inversion recovery (DIR), optimálně v 3D [22]. Ložiska v bílé hmotě nemají tvar Dawsonových prstů, ovoidní tvar ložisek při perivenulární demyelinizaci je u NMOSD méně častý. Práce Jurynczyka et al se věnuje MR kritériím distribuce intracerebrálních ložisek, která jsou schopna s vysokou senzitivitou a specificitou odlišit RS od NMOSD [23]. Je nutné si uvědomit, že ne vždy je přítomno rozsáhlé intramedulární ložisko (LETM) typické pro NMO a že naopak u RS, i když v nižším procentu, se také může vyskytnout rozsáhlejší léze. Dále se u NMOSD mohou vyskytnout i kratší léze, a proto byla snaha definovat kritéria diskriminující obě jednotky na MR zobrazení mozkového parenchymu [22,23]. Kritéria obsahovala: 1. ložisko zasahující k postranní komoře a v oblasti temporálního laloku kaudálně, nebo 2. subkortikální ložisko v oblasti U vláken, nebo 3. ložisko charakteru Dawsonových prstů. Tato kritéria byla schopna odlišit RS s 90,9% sensitivitou a 87,1% specificitou od AQP4-IgGpozNMOSD, resp. s 95,2% MOG-IgGpozNMOSD a 87,5% séronegativní NMOSD [22].
Při volumetrických měřeních je u NMO popisována menší celková atrofie mozku a větší šíře kortexu (větší objem než u RS), z regionální atrofie zde nebyla pozorována výraznější atrofie talamu, která je detekovaná typicky u RS [24].
Kdybych shrnula odlišnosti postižení tří základních oblastí u AQP4-IgGpozNMOSD, MOG-IgGpozNMOSD a RS, tak postižení optického nervu je u AQP4-IgGpozNMOSD typicky bilaterální, dlouhé a zasahující do chiasmatu. U MOG-IgGpozNMOSD je obvyklé dlouhé bilaterální postižení postihující přední část zrakového nervu s intraorbitálním edémem a perineurálním enhancementem po podání kontrastní látky. U RS je postižení obvykle jednostranné a kratší. Postižení míchy je u AQP4-IgGpozNMOSD rozsáhlé, dlouhé přes alespoň tři obratlová těla, je lokalizováno centrálně v šedé hmotě a často zasahuje i do periferie (postihuje více než 50 % arey na transverzálních řezech). U MOG-IgG asociované myelitidy je postižení především v oblasti míšního konu nebo v oblasti dolní hrudní míchy a v thorakolumbálním přechodu, také postihuje především šedou hmotu a může zasahovat periferně (postihuje více než 50 % arey na transverzálních řezech). U RS jsou intramedulární ložiska uložená dorzálně a laterálně, častěji je postižen krční úsek, ložiska jsou drobnější [8]. U AQP4-IgG pozNMOSD je případné postižení mozkového parenchymu periependymální, podél komorového systému a v průběhu kortikospinálního traktu (fokální nebo asociující s vazogenním edémem). U MOG-IgGpozNMOSD je postižení mozkového parenchymu méně časté nebo jsou patrná jen nespecifická ložiska v bílé hmotě (subkortikálně nebo v hluboké bílé hmotě). Může být i obdobné jako u ADEM. V diferenciální diagnostice může pomoci to, že častěji než u RS je postižena centrální šeď (talamy a bazální ganglia) a jsou přítomna atypická infratentoriální ložiska (obr. 11). U RS je typické periventrikulární postižení (tvar – Dawsonovy prsty), postižení CC, callososeptální orientace ložisek, kortikální/juxtakortikální ložiska a postižení infratentoriálně. Ložiska mají typicky ovoidní tvar (obr. 11) [8].
Fig. 11. MRI features of lesions in AQP4-IgGposNMOSD, MOG-IgGposNMOSD and MS patients (adapted from [8]).
![Schéma charakteru ložisek na MR diskriminující AQP4-IgGpozNMOSD, MOG-IgGpozNMOSD a RS (adaptováno z [8]).<br>
Fig. 11. MRI features of lesions in AQP4-IgGposNMOSD, MOG-IgGposNMOSD and MS patients (adapted from [8]).](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image_pdf/054e03c9ea786609153927f2cf5b0df9.png)
AQP4 – aquaporin-4; MOG-IgG – myelin oligodendrocyte glycoprotein antibody; NMOSD – neuromyelitis optica spectrum disorder
Závěr
Nález na MR míchy či MR optických nervů relativně dobře diskriminuje NMOSD od RS či dalších onemocnění postihujících bílou hmotu. Tento nález však nemusí být vždy vyjádřen. V případě postižení míchy se může jednat o drobnější ložisko. Potom narůstá význam podrobného vyhodnocení nálezu na MR mozku a pátrání po ložiskové změně signálu především periependymálně, v oblasti zadního kmene – zvláště v oblasti area postrema a v průběhu pyramidové dráhy.
Grantová podpora
Práce byla podpořena ministerstvem zdravotnictví, výzkumným záměrem RVO 64165 a grantem NV 18-04-00168.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
prof. MUDr. Manuela Vaněčková, Ph.D.
Radiodiagnostická klinika
1. LF UK a VFN v Praze
Kateřinská 30
128 08 Praha 2
e-mail: manuela.vaneckova@vfn.cz
Sources
1. Wingerchuk DM, Weinshenker BG. Neuromyelitis optica. Clinical predictors of a relapsing coure and survival. Neurology 2003; 60 (5): 848–853. doi: 10.1212/01.wnl.0000049912.02954.2c.
2. Vaněčková M, Seidl Z. Roztroušená sklerózy a onemocnění bílé hmoty v MR zobrazení. Praha: Mladá fronta 2018: 286.
3. Wingerchuk DM, Lennon VA, Pittock SJ et al. Revise diagnostic criteria for neuromyelitis optica. Neurology 2006; 66 (10): 1485–1489. doi: 10.1212/01.wnl.0000216139.44259.74.
4. Wingerchuk DM, Banwell B, Cabre P et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015; 85 (2): 177–189. doi: 10.1212/WNL.0000000000001729.
5. Wang KY, Chetta J, Bains P et al. Spectrum of MRI brain lesion patterns in neuromyelitis optica spectrum disorder: a pictorial review. Br J Radiol 2018; 91 (1086): 20170690. doi: 10.1259/bjr.20170690.
6. Kim HJ, Paul F, Lana-Peixoto MA et al. MRI characteristics of neuromyelitis optica spectrum disorder. Neurology 2015; 84 (11): 1165–1173. doi: 10.1212/WNL.0000000000001367.
7. Huh SY, Min JH, Kim W el al. The usefelness of brain MRI at onset in the defferentiation of multiple sclerosis and seropositive neuromyelitis optica spektrum disorders. Mult Scler 2014; 20 (6): 695–704. doi: 10.1177/1352 58513506953.
8. Dutra BG, Rocha AJ, Nunes RH et al. Neuromyelitis optica spectrum disorders: spectrum of MR imaging findings and their differential diagnosis. RadioGraphics 2018; 38 (1): 169–193. doi: 10.1148/rg.2018170141.
9. Pekcevik Y, Orman G, Lee IH et al. What do we known about brain contrast enhancement patterns in neuromyelitis optica? Clin Imaging 2016; 40 (3): 573–580. doi: 10.1016/j.clinimag.2015.07.027.
10. Vaněčková M, Horáková D, Havrdová E et al. Retrospektivní studie nálezů na magnetické rezonanci míchy a mozku u pacientů s diagnózou neuromyelitis optica. Cesk Slov Neurol N 2010; 73/106 (2): 164–168.
11. Jauris S, Paul F, Franciotta D. Mechanisms of disease: aquaporin-4 antibodies in neuromyelitis optica. Nat Clin Pract Neurol 2008; 4 (4): 202–214. doi: 10.1038/nc pneuro0764.
12. Pekcevik Y, Mitchell CH, Mealy MA et al. Differentiating neuromyelitis optica from other causes of longitudinally extensive transverse myelitis on spinal magnetic resonance imaging. Mult Scler 2016; 22 (3): 302–311. doi: 10.1177/1352458515591069.
13. Huh SY, Kim SH, Hyun JW et al. Short segment myelitis as a first manifestation of neuromyelitis optica spectrum disorders. Mult Scler 2017; 23 (3): 413–419. doi: 10.1177/1352458516687043.
14. Flanagan EP, Weinshenker BG, Krecke KN et al. Short myelitis lesions in aquaporin-4.IgG-positive neuromyelitis optica spectrum disorders. JAMA Neurol 2015; 72 (1): 81–87. doi: 10.1001/jamaneurol.2014.2137.
15. Ramanathan S, Prelog K, Barnes EH et al. Radiological differention of optic neuritis with myelin oligodendrocyte glycoprotein antibodies, aquaporin-4 antibodies, and multiple sclerosis. Mult Scler J 2016; 22 (4): 470–482. doi: 10.1177/1352458515593406.
16. Wynford-Thomas R, Jacob A, Tomassini V. Neurological update: MOG antibody disease. J Neurol 2019; 266 (5): 1280–1286. doi: 10.1007/s00415-018-9122-2.
17. Deneve M, Biotti D, Patsoura S et al. MRI features of demyelinating disease associated with anti-MOG antibodies in adults. J Neuroradiol 2019; 46 (5): 312–318. doi: 10.1016/j.neurad.2019.06.001.
18. Jarius S, Ruprecht K, Kleiter I et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part1: fequency, syndrome specificity, influence of disease aktivity, long-term course, association with AQP4-IgG and origin. J Neuroinflammation 2016; 13 (1): 279. doi: 10.1186/s12974-016-0717-1.
19. Cobo-Calvo A, Ruiz A, Maillart E et al. Clinical spectrum and prognostic value of CNS MOG autoimunity in adults: the MOGADOR study. Neurology 2018: 90 (21): e1858–e1869. doi: 10.1212/WNL.0000000000005560.
20. Ikeda A, Watanabe Y, Kaba H et al. MRI findings in pediatric neuromyelitis optica spectrum disorder with MOG antibody: four cases and review of the literature. Brain Dev 2019; 41 (4): 367–372. doi: 10.1016/j.braindev.2018.10.011.
21. Fernandez-Carbonell C, Vargas-LowyD, Musallam A et al. Clinical and MRI phenotype of children with MOG antibodies. Mult Scler 2016; 22 (2): 174–184. doi: 10.1177/1352458515587751.
22. Eichinger P, Schön S, Pongratz V et al. Accuracy of unenhanced MRI in the detection of new brain lesions in multiple sclerosis. Radiology 2019; 291 (2): 429–435. doi: 10.1148/radiol.2019181568.
23. Jurynczyk M, Tackley G, Kong Y et al. Brain lesion distribution criteria distinguish MS from AQP4-antibody NMOSD and MOG-antibody disease. J Neurol Neurosurg Psychiatry 2017; 88 (2): 132–136. doi: 10.1136/jnnp-2016-314005.
24. Duan Y, Liu Y, Liang P et al. Comparision of grey matter atrophy between patients with neuromyelitis optica and multiple sclerosis: a voxel-based morphometry study. Eur J Radiol 2012; 81 (2): e110–e114. doi: 10.1016/j.ejrad.2011.01.065.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2020 Issue supplementum 1
Most read in this issue
- Magnetic resonance imaging in neuromyelitis optica spectrum disorders
- Neuromyelitis optica spectrum disorders – laboratory examination
- Epidemiology, clinical manifestation, and disease course of neuromyelitis optica spectrum disorders
- Differential diagnosis of neuromyelitis optica spectrum disorders