
Leberova hereditární neuropatie optiku
Leber Hereditary Optic Neuropathy
Leber hereditary optic neuropathy (LHON) is maternally inherited disorder characterized by subacute loss of vision due to impairment of retinal ganglion cells eventually leading to optic atrophy. Three prevalent point mutations in mitochondrial DNA: m.11778G>A, m.3460G>A, and m.14484T>C, are causative in the majority (95%) of cases. All of these mutations affect one of the subunits of complex I, NADH-ubiquinone oxidoreductase, the first enzyme of the mitochondrial respiratory chain. The presence of a mutation is necessary but not sufficient to cause visual loss. The penetrance is incomplete with only about 50% of men and 10% of women event. developing clinical signs of the disease. Recently, it was proven that early initiation of therapy with idebenone in patients manifesting LHON ameliorates visual functions and clinical trials testing several other promising therapies are underway. Incomplete penetrance, similarites with other disorders affecting the optic nerve and a great variability of clinical features cause considerable diagnostic difficulties. Often there is a delay in therapy initiation, as late as in the phase of the irreversible optic nerve damage. In 2013, we established a multidisciplinary medical care centre dedicated to patients with mitochondrial optic neuropathies in General University Hospital in Prague to develop effective diagnostic and treatment algorithms and to study the underlying pathogenetic mechanisms.
Key words:
LHON – optic neuropathy – optic atrophy – mitochondria – mtDNA – multiple sclerosis
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
:
H. Kolářová 1; T. Honzík 1; Ľ. Ďuďáková 2; B. Kousal 3; J. Kulhánek 1; P. Diblík 3; M. Tesařová 1; P. Havránková 4; M. Forgáč 4; J. Zeman 1; P. Lišková 2,3
:
Klinika dětského a dorostového lékařství 1. LF UK a VFN v Praze
1; Ústav dědičných metabolických poruch, 1. LF UK a VFN v Praze
2; Oční klinika 1. LF UK a VFN v Praze
3; Neurologická klinika 1. LF UK a VFN v Praze
4
:
Cesk Slov Neurol N 2017; 80/113(5): 534-544
:
Review Article
prolekare.web.journal.doi_sk:
https://doi.org/10.14735/amcsnn2017534
Leberova hereditární neuropatie optiku (LHON) je maternálně dědičné onemocnění projevující se rychlou, nebolestivou ztrátou zraku v důsledku poruchy funkce a odumírání retinálních gangliových buněk, což v konečném důsledku vede k atrofii zrakového nervu. Více než 95 % pacientů nese jednu ze tří prevalentních bodových mutací v genech mitochondriální DNA: m.11778G>A, m.3460G>A a m.14484T>C. Všechny tři mutace zasahují některou z podjednotek komplexu I, NADH-koenzym Q10 oxidoreduktázy, prvního enzymu z mitochondriálního respiračního řetězce. Přítomnost mutace je nezbytný, ale nikoliv postačující patofyziologický prvek postižení zrakových funkcí, jak je patrno z neúplné penetrance onemocnění. Zcela nedávno bylo prokázáno, že včasné zahájení terapie idebenonem u pacientů po manifestaci LHON vede k významnému zlepšení výsledných zrakových funkcí, a další možnosti terapie jsou ve fázi klinických zkoušek. Neúplná penetrance, rychlý rozvoj onemocnění a velká variabilita klinického obrazu vedou ke značným diagnostickým obtížím, které mají často za následek pozdní zahájení léčby – již ve fázi nevratného poškození zrakového nervu. V roce 2013 jsme v rámci VFN v Praze vytvořili centrum multidisciplinární péče pro pacienty s mitochondriálními neuropatiemi optiku, jehož cílem je optimalizace diagnostických a terapeutických algoritmů a studium rizikových faktorů rozvoje manifestního onemocnění.
Klíčová slova:
LHON – optická neuropatie – optická atrofie – mitochondrie – mtDNA – sclerosis multiplex
Úvod
Postižení oka patří spolu s encefalopatií a myopatií mezi nejběžnější projevy mitochondriální dysfunkce a zahrnuje oftalmoplegii a nebo ptózu, pigmentovou degeneraci sítnice, makulopatie a neuropatie optiku. Leberova hereditární neuropatie optiku (LHON, OMIM 535000) představuje nejčastější příčinu mitochondriálních neuropatií optiku způsobující progresivní ztrátu zraku. Přestože byla mitochondriální onemocnění dříve považována za poměrně vzácná, jejich současná prevalence je, především díky zlepšení diagnostiky, odhadována až na 1 : 5 000 živě narozených dětí [1]. Jedná se o heterogenní skupinu poruch, které se manifestují zejména multisystémovým postižením, ale vzácněji se mohou projevit také izolovanou dysfunkcí jednoho orgánu.
Onemocnění LHON bylo poprvé rozpoznáno německým oftalmologem Albrechtem von Graefem v roce 1858, avšak pojmenováno bylo po jeho asistentovi Theodoru Leberovi, který později popsal klinický průběh onemocnění u 15 pacientů [2]. LHON je první onemocnění, jež bylo asociováno s maternální dědičností a konkrétní bodovou mutací v mitochondriální DNA (mtDNA) [3,4]. Více než 95 % pacientů nese jednu ze tří prevalentních bodových mutací v genech mtDNA: m.11778G>A, m.3460G>A a m.14484T>C (obr. 1) [5]. Všechny tři mutace zasahují některou z podjednotek komplexu I, NADH-koenzym Q10 oxidoreduktázy, prvního enzymu z mitochondriálního respiračního řetězce [6]. Přítomnost mutace je nezbytný, nikoli však jediný patofyziologický prvek postižení zrakových funkcí. Přestože se u mitochondriálních onemocnění dříve předpokládalo, že práh pro manifestaci onemocnění (tzv. trigger point) je asi 80 – 90 % mutovaných mtDNA molekul v buňce, naprostá většina všech symptomatických i asymptomatických jedinců s LHON nese mutace v homoplazmické podobě (stav, kdy jsou v buňce všechny molekuly mtDNA stejné, zde tedy ve 100 % mutované). Předpokládá se, že k rozvoji manifestního onemocnění je nutná souhra několika endogenních i exogenních faktorů.
![Linearizovaná strukturální mapa mitochondriální DNA (převzato z [111]).
Červené šipky označují 3 LHON prevalentní mutace v genech kódujících ND1, ND4 a ND6 podjednotky komplexu I.
Fig. 1. The linearized structural map of the human mtDNA (modified from [111]).
The three prevalent LHON mutations in genes encoding ND1, ND4 and ND6 subunits of the Complex I are marked by arrows.](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image/49ea4bbe0a83359003dfb55184f0fcc0.jpg)
Přesná data o výskytu LHON nejsou, na rozdíl od jiných evropských států, v České republice k dispozici. Prevalence manifestního onemocnění se v Anglii pohybuje okolo 1 : 31 000 (prevalence jedinců s potvrzenoumutací, tzv. asymptomatických nosičů, je však odhadována až na 1 : 8 500) [7,8], 1 : 39 000 v Nizozemí [9] a 1 : 50 000 ve Finsku [10] a 1 : 54 000 v Dánsku. Mezi lety 1992 a 2016 bylo na našem pracovišti diagnostikováno přes 90 jedinců české národnosti s jednou ze tří prevalentních mutací, z čehož 20 bylo symptomatických. Pokud počítáme s obdobnou prevalencí jako na britských ostrovech a Nizozemí, kde je i obdobné mtDNA haploskupinové rozložení, mělo by u nás být odhadem přes 300 asymptomatických nosičů a 80–90 pacientů s manifestním onemocněním. Je tedy zřejmé, že onemocnění je v České republice značně poddiagnostikováno.
Lékaři prvního kontaktu s pacienty majícími LHON jsou nejčastěji oční lékař nebo neurolog. Vzhledem k tomu, že toto onemocnění bývá často nesprávně vyhodnoceno a následně i léčeno jako roztroušená skleróza (RS) či neuromyelitis optica (NMO) neboli morbus Devic, od kterých je v první fázi prakticky neodlišitelné, dochází často k opožděnému zahájení správné terapie, která pak nedosahuje odpovídajících výsledků. Prostřednictvím tohoto souhrnného pojednání představujeme základní charakteristiky a specifika onemocnění LHON.
Klinický průběh onemocnění
Klinický průběh LHON obvykle zahrnuje rychlou nebolestivou ztrátu zrakové ostrosti obou očí během několika dnů až týdnů (subakutní průběh). K rozvoji klinických příznaků dochází obvykle mezi 15 a 35 roky, přičemž u 90 % pacientů se onemocnění projeví do 50 let věku [11]. Rozvoj poruchy zrakových funkcí se však může objevit i v průběhu první dekády života [12,13] či velmi zřídka až po 70.–80. roce věku [14]. Muži mají 4–5× větší pravděpodobnost, že se u nich choroba projeví. Pohlaví ani příčinné mutace ovšem nemají vliv na to, kdy onemocnění vznikne a jak závažná bude ztráta zrakových funkcí při manifestaci [11].
Obvykle dochází během 4–6 týdnů na jednom oku k progresivnímu snižování zrakové ostrosti a výrazné poruše barvocitu a kontrastní citlivosti. První obvykle bývají zasažena nejtenčí vlákna papilomakulárního svazku, což se projevuje jako vznik centrocekálních skotomů (obr. 2A, B) [5,15]. Zornicové reflexy zůstávají v porovnání s rozsahem ztráty zraku relativně zachovány. To má význam v diferenciální diagnostice tohoto onemocnění [16,17]. Stejné symptomy se po průměrné době 6–8 týdnů rozvinou na oku druhém [13]. U naprosté většiny pacientů jsou postupně postiženy obě oči během 6 měsíců [11]. Asi ve 25 % případů dochází ke ztrátě zrakových funkcí současně u obou očí již na začátku onemocnění [11].

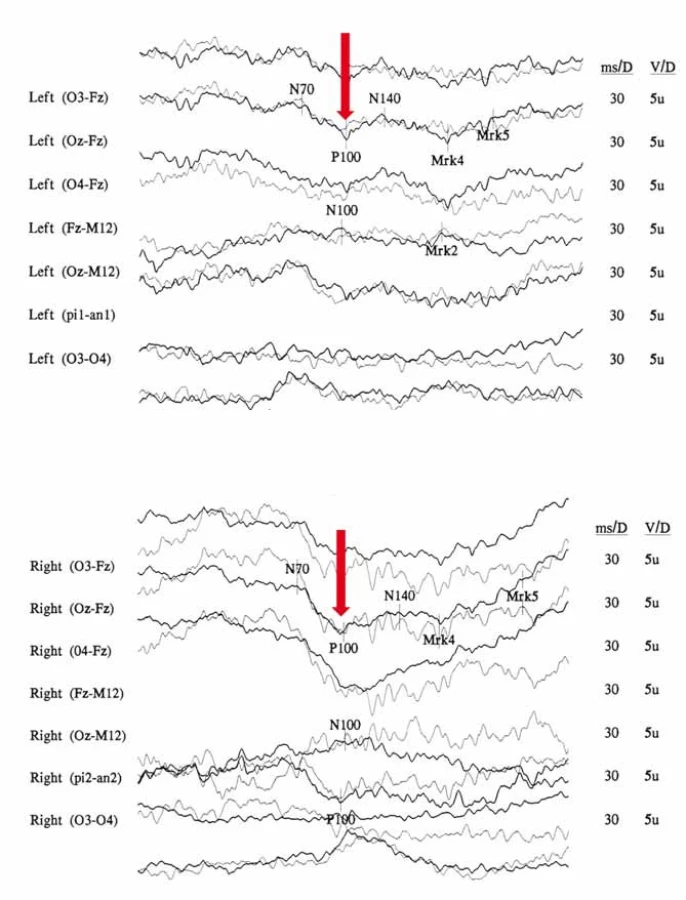
Na očním pozadí bývá v akutní fázi patrné prosáknutí papily zrakového nervu s rozšířením a zvýšenou vinutostí cirkumpapilárních cév (teleangiektatická mikroangiopatie) (obr. 3A). Při fluorescenční angiografii však nedochází k úniku barviva, což vytváří obraz tzv. pseudoedému [20]. Ztluštění vrstvy retinálních nervových vláken (RNFL) v okolí disku lze objektivizovat např. pomocí optické koherenční tomografie se spektrální doménou (SD-OCT) (obr. 3B). Při vyšetření zrakových evokovaných potenciálů (VEP) a elektroretinografie (ERG) je prokazováno v akutní fázi snížení amplitud a prodloužení latencí kortikálních odpovědí (obr. 4A, B) [18]. Je třeba zdůraznit, že přibližně 20 % pacientů má na očním pozadí normální nález, což může vést k nesprávné diagnóze funkční poruchy zraku [12,13,19,20].
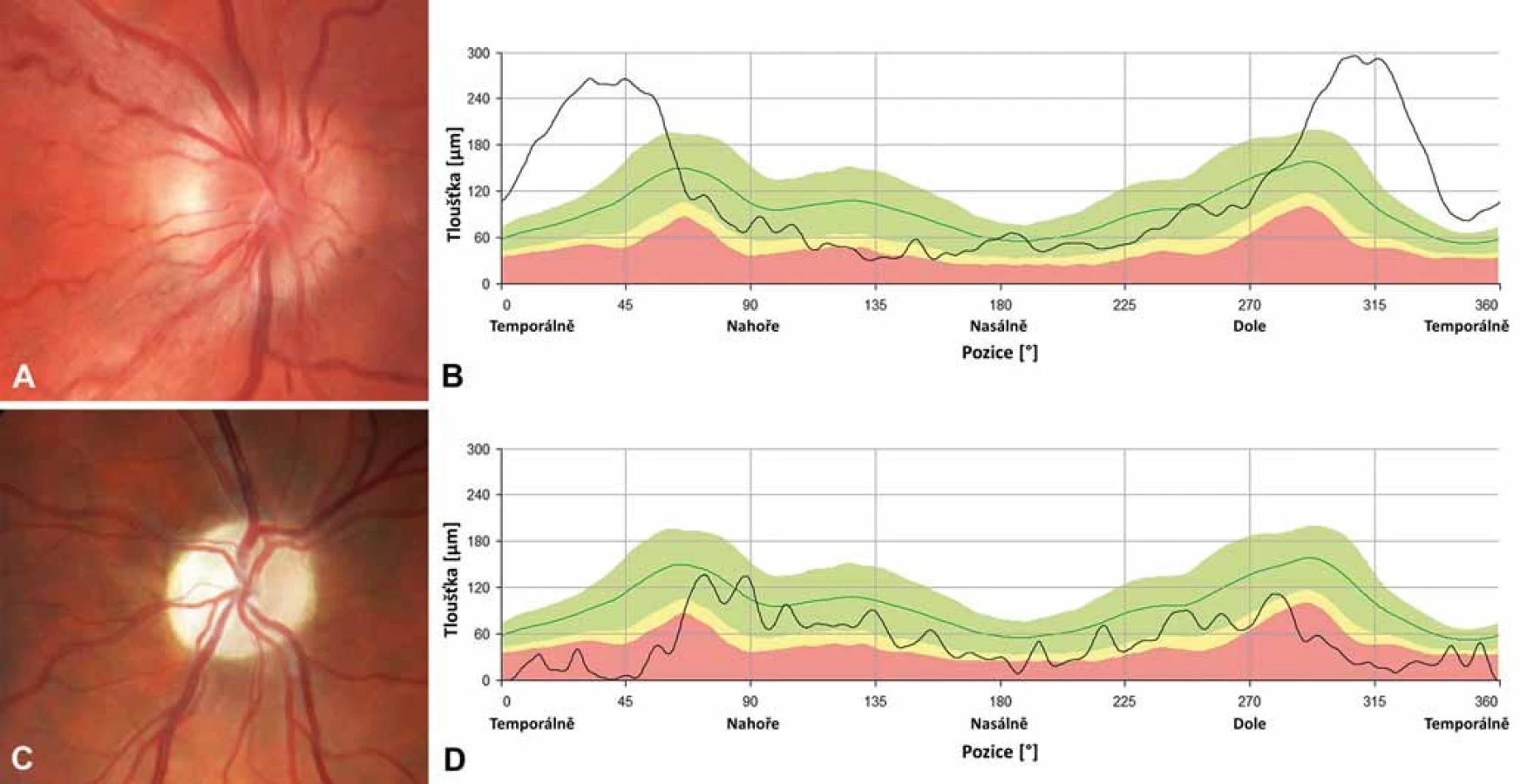
Během 6 týdnů od manifestace se zrakové funkce stabilizují a onemocnění přechází do chronické fáze na podkladě progresivní atrofie zrakového nervu, kterou lze objektivizovat průkazem ztenčování RNFL [21]. K rozvoji kompletní atrofie dochází minimálně po dobu 12 měsíců [22], nejprve v oblasti temporálního a dolního kvadrantu a následně i v kvadrantu horním a nazálním, který je někdy relativně ušetřen (obr. 3C, D) [15,21]. Postižení zrakových funkcí je závažné – u většiny pacientů vznikají rozsáhlé centrální skotomy (obr. 2C, D) a zraková ostrost je významně snížená až do pásma těžké slabozrakosti tj. ≤ 0,1 dle WHO (odpovídá 3/60–1/60, resp. 0,05–0,02 na Snellenových optotypech a 1,0 logMAR), či praktické slepoty (1,3 logMAR; ≤ 1/60, resp. 0,02 [23].
Po odeznění akutní fáze popisují některé studie možné zlepšení zrakových funkcí, a to častěji u mladších pacientů (< 10 let), kteří mívají akutní fázi prodlouženou s pomalejším úbytkem zrakových funkcí a u pacientů s relativně velkými terči zrakového nervu [24,25]. Je známo, že i mezi jednotlivými mutacemi existují rozdíly v penetranci onemocnění a závažnosti projevů, především konečného stavu zrakových funkcí (tab. 1) [26]. Nejčastěji je spontánní zlepšení popisováno u mutace m.14484T>C (ve 37 – 64 %) [9,27,28]. Oproti tomu u m.3460G>A je podíl pacientů s návratem zrakových funkci jen 15–25 % [9,20,29] a u m.11778G>A pouze 4–25 % [20,30,31].

Výjimečně může u některých jedinců nesoucích LHON prevalentní mutaci vzniknout pouze mírné postižení funkce zrakového nervu, které se projevuje jako porucha barvocitu s neschopností rozlišovat nejčastěji červenou a zelenou barvu, snížení kontrastní citlivosti a lehce abnormální ERG a VEP [32].
Je zajímavé, že abnormality při vyšetření fundu a vyšetření sítnice pomocí SD-OCT (např. cirkumpapilární teleangiektatická mikroangiopatie, ztluštění RNFL) byly pozorovány také u jedinců v presymptomatickém stadiu nemoci, a dokonce u asymptomatických nosičů mutace, u kterých se nikdy nerozvinul klinický obraz ztráty zrakových funkcí [12,33]. Často se jedná pouze o diskrétní změny; během života však může docházet k jejich minimalizaci až úplnému vymizení nebo naopak k jejich zvýraznění [34]. Subklinické odchylky byly pozorovány i v rámci elektrofyziologických vyšetření VEP a ERG [34,35]. Livingstone et al (1980) studovali 16 asymptomatických nosičů. U jednoho nalezli alterace při vyšetření VEP a zvažovali, zda se může jednat o časnou známku rozvoje onemocnění [36]. Signifikantní prodloužení vlny P100 při vyšetření VEP bylo prokázáno i ve skupině 48 asymptomatických nosičů mutace m.11778G>A, u kterých nikdy nevznikla trvalá porucha zraku [35]. Je tedy více než pravděpodobné, že strukturní změny nekorelují s klinickým fenotypem, a prediktivni hodnota vyšetření VEP i dalších funkčních i zobrazovacích vyšetření proto stále zůstává nejistá.
Jednostranné postižení zrakového nervu je u LHON extrémně vzácné, v těchto případech je třeba aktivně hledat a vyloučit jiné patologické procesy [11]. Popsáno bylo u dětí, nicméně i v těchto případech bylo spojeno buď se subklinickým postižením oka druhého, nebo s manifestací na druhém oku v časné dospělosti po dlouhém asymptomatickém intervalu [24] (osobní komunikace, prof. Carelli).
Extraokulární manifestace
Přestože se LHON nejčastěji projevuje jako izolované postižení zrakového nervu, v některých případech se onemocnění může komplikovat i dalšími doprovodnými příznaky, jakými jsou např. periferní neuropatie, myopatie, převodní poruchy srdečního rytmu a dyskineze končetin [37]. Stále však není jasné, zda jejich, obvykle velmi izolovaný, výskyt nepředstavuje jen pouhou koincidenci. Vzácněji byly u pacientů pozorovány neurologické projevy závažnějšího charakteru, jako např. spastická dystonie, ataxie, juvenilní encefalopatie a psychiatrické poruchy, které bývají označovány jako LHON plus nebo jako překryvné (tzv. overlapping) syndromy. Jejich výskyt bývá vázán na jiné mutace ovlivňující aktivitu komplexu I či jde o ojedinělé případy z geograficky omezených oblastí [38–45].
Výše bylo uvedeno, že odlišení LHON od RS v akutní fázi onemocnění činí značné diagnostické obtíže. Tento fakt se dále komplikuje stále neobjasněnou otázkou možné asociace LHON s RS (tzv. syndrom Hardingové) [46]. U naprosté většiny pacientů s LHON je v akutní fázi onemocnění obraz na MR bez patologického nálezu [47]. V atrofické fázi mohu být na MR naopak patrny hypersignality optického nervu na podkladě gliózy [48]. Část pacientů s LHON však rozvíjí klinický i radiologický obraz neodlišitelný od RS s nálezem charakteristických lézí bílé hmoty v periventrikulárních oblastech a oligoklonálních IgG pásů při vyšetření mozkomíšního moku [49 – 51]. Pacienti se manifestují opakovanou poruchou zrakových funkcí spojenou s bolestí za okem hlavně při jeho pohybu [52]. V těchto případech obvykle nedochází ke kompletní obnově zrakových funkcí po nasazení terapie kortikosteroidy a v polovině případů u pacientů porucha progreduje do praktické slepoty [52]. Druhá skupina pacientů s LHON však má pouze radiologické známky RS a otázka jejich budoucí manifestace zatím zůstává nejasná [53]. Někteří autoři uvádějí až 50× vyšší riziko rozvoje RS ve skupině LHON pacientů, a to především u symptomatických žen [13,46,51–54]. Pokud se tedy žena s LHON manifestuje ztrátou zraku, lze očekávat závažnější průběh neurologického postižení než u mužů. Existují teorie, které naznačují, že primární mtDNA mutace může spouštět autoimunitní odpověď vedoucí k demyelinizaci axonů a rozvoji RS [46,51]. Další možným vysvětlením je, že velké neurologické poškození při RS může vést k sekundárnímu mitochondriálnímu poškození, a podmiňovat tak rozvoj LHON [53]. V současnosti nepanuje v otázce vyššího výskytu RS ve skupině pacientů s LHON shoda, neboť část odborníků je přesvědčena, že se jedná o pouhé populační riziko [52].
Diagnostika
Podezření na onemocnění LHON často vyslovuje již oční lékař nebo neurolog, a to na základě správného odebrání anamnézy a zhodnocení podrobného očního vyšetření spočívajícího ve vyšetření zrakové ostrosti, zorného pole, kontrastní citlivosti, barvocitu, popř. RNFL. Zlatým standardem laboratorní diagnostiky je posléze molekulárně genetická analýza prevalentních mutací ze vzorků krve či z buněk stěru bukální sliznice. Toto vyšetření se provádí u pacientů s již rozvinutou poruchou zraku v rámci diferenciální diagnostiky LHON či u rodinných příslušníků, kteří zatím nemají klinické obtíže, a kteří se tak vyhnou náročnému diagnostickému procesu. Molekulárně genetické vyšetření však u těchto asymptomatických pacientů neumožňuje predikovat rozvoj onemocnění. Při vyloučení prevalentních mutací je vhodné zvážit sekvenaci mtDNA genů kódujících podjednotky komplexu I v mitochondriích izolovaných ze svalové biopsie.
Patogeneze onemocnění
Přestože je LHON vzhledem k četnosti jeho výskytu jedno z nejvíce studovaných mitochondriálních onemocnění, mnoho důležitých otázek stále zůstává neobjasněných. Proč jsou u tohoto onemocnění postiženy prakticky výlučně RGB tvořící zrakový nerv? Proč se v jedné rodině nesoucí stejnou a ve většině případů homoplazmickou mutaci projeví onemocnění jen v některých případech? Jaké je vysvětlení pro někdy až osminásobně vyšší výskyt symptomatického onemocnění u mužů (tab. 1)? Souhrnný výčet známých faktorů ovlivňujících penetranci onemocnění je uveden ve schématu na obr. 5.
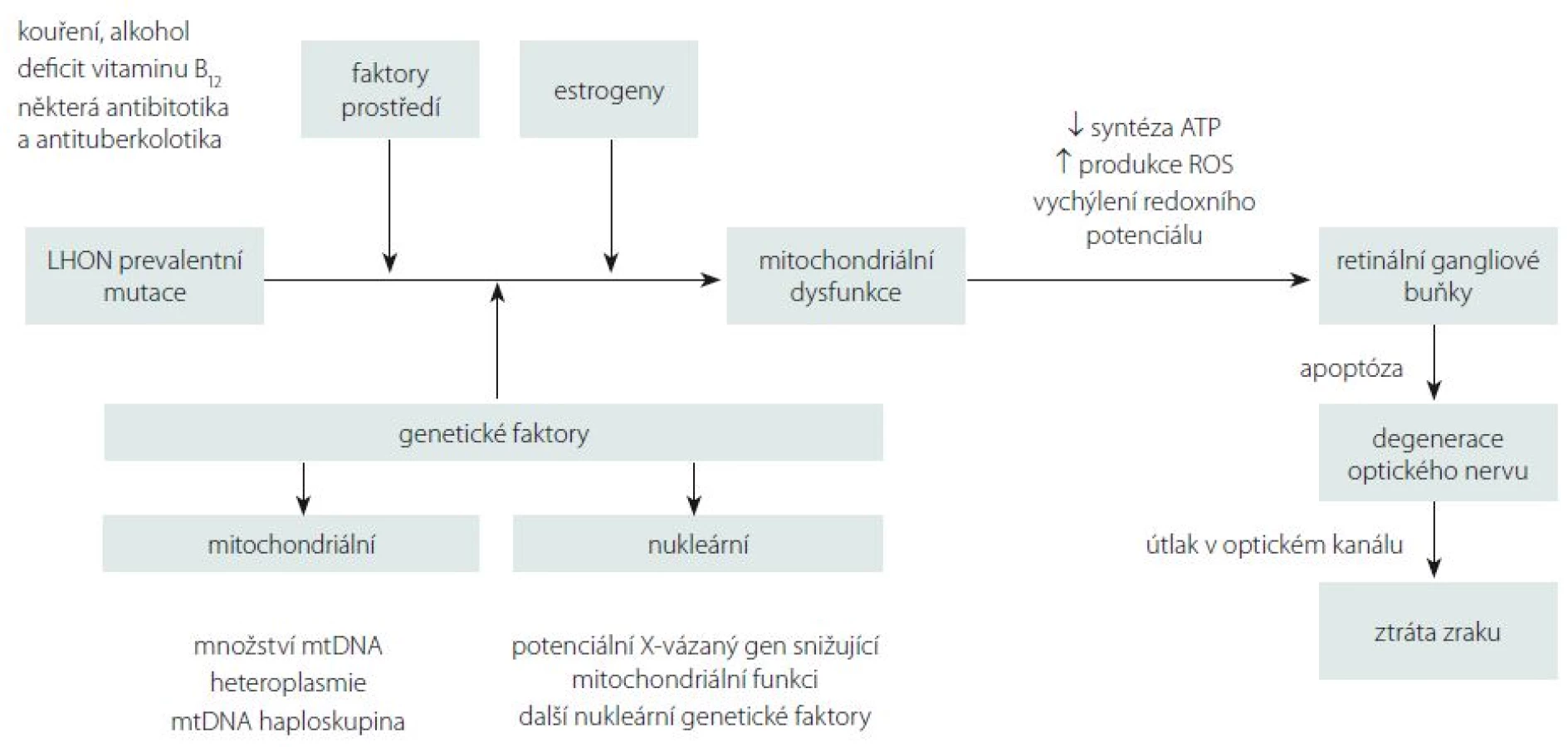
Jedním z možných vysvětlení selektivního postižení RGB jsou jejich vysoké nároky na nepřetržitou dodávku ATP (adenosintrifosfát). Histochemickými studiemi bylo prokázáno zvýšené nahromadění mitochondrií v oblasti lamina cribrosa sclerae, kde ještě nemyelinizovaná nervová vlákna opouští sítnici, aby vytvořila zrakový nerv [5]. Tato oblast je bohatá na enzymy ze skupiny Na+/K+ ATPáz, což činí z místního neuronálního vedení energeticky nesmírně náročný proces a může vysvětlit výjimečnou zranitelnost prelaminárních vláken zrakového nervu [55]. Defekt mitochondriálního metabolizmu má za následek lokální stázu axoplazmy s otokem, který dále přispívá k degeneraci vrstvy RGB a jejich axonů tvořících zrakový nerv [56]. Proti této teorii stojí fakt, že fotoreceptory, které jsou u LHON zachovány, mají mnohem vyšší oxidativní nároky než RGB [57,58]. Navíc jiná mitochondriální onemocnění se závažnější poruchou komplexu I nevedou vždy k rozvoji atrofie optiku [6]. Je proto možné, že spíše než na nedostatek ATP jsou RGB citlivé na drobná vychýlení buněčného redoxního potenciálu a produkci kyslíkových radikálů (Reactive Oxygen Species; ROS) [59].
Klinický i patogenetický obraz LHON se částečně překrývá s obrazem dominantní atrofie optiku (DOA, OMIM 165500) na podkladě mutací v nukleárním genu OPA1. Tento gen kóduje protein ze skupiny dynaminů, který je odpovědný za udržování integrity mitochondriálního genomu, tvorbu krist a fúzi mitochondrií [60,61]. Zdá se proto, že správná funkce RGB je závislá na zachovalé biogenezi mitochondrií. Giordano et al ve své studii prokázali, že množství mtDNA u LHON negativně koreluje s rozvojem axonálního otoku a dalších subklinických změn u pacientů s LHON, a vyslovili hypotézu, že zvýšená mitochondriální biogeneze na podkladě replikace mtDNA může být významný protektivní faktor rozvoje manifestního onemocnění [62]. Na základě této teorie by k rozvoji manifestního onemocnění došlo pouze u těch nosičů mutace, kteří ztratí schopnost zvýšit svou mitochondriální biogenezi. Stanovení množství mtDNA by tak v budoucnu mohlo sloužit jako významný biomarker prognózy onemocnění [62]. Dosud však nejsou k dispozici longitudinální data, která by tuto teorii potvrdila.
Sadun et al udávají, že svou roli v penetranci onemocnění hrají i další nukleární či mitochondriální genetické faktory [63]. Výše byly uvedeny rozdíly v závažnosti projevů a penetranci onemocnění mezi třemi prevalentními mutacemi [13]. Přídatný negativní efekt na prevalentní mutaci a rozvoj klinicky manifestního onemocnění mohou mít přidružené mtDNA polymorfizmy. Během evoluce došlo k jejich segregaci v tzv. haploskupinách, které se následně dědily společně. V Evropě bylo popsáno celkem 11 haploskupin [64]. Na základě fylogenetických studií byla prokázána preferenční asociace prevalentních mutací LHON s haploskupinou J přítomnou u 9 % evropské populace [65–67]. V České republice, která má obdobné haploskupinové rozložení jako zbytek Evropy, je její frekvence odhadována na 8 %. [64]. Buněčné linie nesoucí mutaci m.11778G>A a haploskupinu J měly prokazatelně nižší spotřebu kyslíku v porovnání s liniemi nesoucími pouze mutaci [68]. Největší asociace s touto haploskupinou byla prokázána u m.14484T>C (80 %) [69]. Naopak m.3460G>A se zdá být mezi jednotlivými haploskupinami distribuována víceméně náhodně [70], což by dále potvrzovalo její závažnost a vysokou penetranci bez nutnosti působení přídatných rizikových faktorů [71].
Přestože naprostá většina pacientů s LHON má mutace v homoplazmické podobě, 10–15 % mutací je heteroplazmických a tkáňově specifická segregace může být odpovědná za rozdílný interindividuální fenotyp [6,20,72]. Některé studie naznačují, že u pacientů je riziko minimální, pokud je heteroplazmie nižší než 60 % [73]. Matky, které mají hladinu heteroplazmie rovnou nebo nižší 80 %, mají menší pravděpodobnost, že u jejich synů dojde k manifestaci onemocnění [73]. Kazuistiky dvou symptomatických pacientů s 15% a 26% hladinou heteroplazmie v leukocytech ale spíše poukazují na její nízkou korelaci s fenotypem [74,75] nebo na odlišnou hladinu heteroplazmie v různých tkáních. Studie Howella et al popsala 33% hladinu heteroplazmie v leukocytech periferní krve v porovnání s 95%, resp. 100% hladinou heteroplazmie ve zrakovém nervu a sítnici [76].
Prevalence heteroplazmie je nejvyšší u m.3460G>A (40 %) oproti 36,4 % u m.14484T>C a 5,6 % u m.11778G>A [75]. Heteroplazmie pacientů s LHON se navíc může v čase měnit; u m.3460G>A mutace segregační analýza leukocytů periferní krve prokázala genetický drift vedoucí k vyšší frekvenci mutovaných alel u následujících generací [72,75,77], ale naopak možnou negativní selekci wild-type alel hematopoetických buněk v průběhu života [78]. Kromě toho existují teorie, které naznačují, že heteroplazmie je pouze přechodné stadium od jednoho homoplazmického (wild-type) stadia ke stadiu homoplazmickému (mutovanému) [75]. Výskyt heteroplazmie v konkrétní rodině by tak naznačoval, že se mutace odehrála relativně nedávno [75]. Dle této teorie by i mutace m.3460G>A, která má největší procento heteroplazmických případů mohla být fylogeneticky nejmladší.
Velmi diskutovanou otázkou je i výskyt LHON u ženských nositelek mutace, které mají v závislosti na konkrétní genetickém pozadí jednoznačně nižší penetranci než muži (tab. 1). Některé studie naznačují, že za rozdílnou penetrancí by mohl stát modifikující X-vázaný gen, který povede k manifestaci u žen pouze v homozygotním stavu či X-inaktivací wild-type X chromozomu [79]. Do dnešního dne bylo popsáno několik možných lokusů, které by mohly zvyšovat náchylnost ke vzniku manifestního onemocnění [20,80]; konkrétní gen však zatím nebyl potvrzen. Kromě toho nebyla v leukocytech postižených žen sešikmená inaktivace X chromozomu nalezena [81–84]. Velmi diskutovanou otázkou je i protektivní úloha estrogenů (obr. 5). Zdá se, že zvýšeného kompenzačního potenciálu je u žen docíleno několika mechanizmy jejich působení: aktivací antioxidačního enzymu superoxiddismutázy 2, zvýšením mitochondriální biogeneze a malým, ale prokazatelným zvýšením buněčné energetické kapacity [85]. Těchto poznatků může být využito v potenciálním terapeutickém ovlivnění estrogenového receptoru β (ER-β), hojně přítomném v RGB [85].
V neposlední řadě je nutno zmínit úlohu faktorů prostředí, které mohou mít vliv na manifestaci onemocnění. Na prvním místě stojí nepochybně kouření. Studie 206 asymptomatických nositelů mutace prokázala 93 % penetranci u mužů kuřáků [23]. Ve stejné studii byl potvrzen pouze statisticky nevýznamný trend zvýšené manifestace onemocnění u pacientů konzumujících nadměrné množství alkoholu, přesto je vzhledem k závažnosti klinických projevů rozumné pacientům doporučit, aby omezili nadměrné, a především nárazové pití alkoholu [23]. Jako další možný rizikový faktor rozvoje manifestního onemocnění je uváděn i nedostatek vitaminu B12 [86,87]. Ve vztahu k endosymbiotické teorii poukazující na bakteriální původ mitochondrie je na zvážení užití antibiotik a antituberkulotik interferujících s jejich proteosyntetickým aparátem (makrolidy, aminoglykosidy, etambutol, izoniazid a linezolid) [23,37]. Na základě našich zkušenosti si ale nemyslíme, že by standardní 2–3týdenní terapie antibiotiky vedla k rozvoji manifestního onemocnění.
Diferenciální diagnostika
Jakýkoli zásah poškozující RGB a nebo jejich axony může mít za následek rozvoj neuropatie optiku. Atrofie zrakového nervu mohou vzniknout na podkladě ischemických, infiltrativních, toxických, nutričních, útlakových a traumatických procesů probíhajících v oblasti orbity, chiazmatu či průběhu tractus opticus. V diferenciální diagnostice je dále nutno odlišit refrakční vady (astenopické obtíže) a psychosomatické obtíže způsobující tzv. funkční poruchy zraku.
Hlavní diagnostické rozpaky způsobuje především několik demyelinizačních zánětlivých onemocnění, které na rozdíl od LHON většinou odpovídají na terapii kortikosteroidy. Patří mezi ně především RS, NMO a chronická recidivující zánětlivá neuropatie optiku (Chronic Relapsing Inflammatory optic Neuropathy; CRION) [88]. Oproti LHON se RS vyskytuje mnohem častěji u žen. Ztráta zraku bývá obvykle pouze jednostranná a doprovází ji tlak až bolesti za okem při jeho pohybech [89]. Také progrese zrakové dysfunkce bývá spíše akutního rázu (1 – 3 dny) a většinou nebývá tak závažná jako u LHON [89]. Podobně se manifestuje i jiné, vzácnější autoimunitní onemocnění, kterým je CRION [90]. Jak retrobulbární neuritida v rámci RS, tak CRION mají oproti LHON často porušeny zornicové reflexy [91]. NMO byla dříve považována za formu RS [92,93]. Příčina tohoto onemocnění byla prokázána v tvorbě protilátek proti akvaporinu 4 (AQP4-IgG), které po navázání na cílový antigen spouštějí komplementovou kaskádu a lokální zánětlivou reakci se všemi jejími důsledky [94,95]. Vyšetření těchto protilátek může pomoci v diferenciální diagnostice těchto onemocnění [96].
Hlavní hereditární neuropatií optiku, kterou je nutno vyloučit v rámci diferenciální diagnostiky LHON, je DOA, jež nejčastěji vzniká na podkladě mutací v OPA1. DOA postihuje ve stejném poměru ženy i muže a nejčastěji se manifestuje během prvních dvou dekád života bilaterální poruchou zrakové ostrosti a barvocitu. Oproti LHON je progrese onemocnění pomalá s rozdílnou prognózou, od prakticky asymptomatického průběhu po rozvoj praktické slepoty [97].
Terapie
Terapeutické možnosti LHON jsou stále relativně omezeny. V současné době neexistuje léčba, která by dokázala předejít manifestaci onemocnění či plně navrátit zrakové funkce. V chronickém stadiu onemocnění je pro pacienty stěžejní terapie symptomatická či podpůrná za použití speciálních optických (např. lupy či monokuláry) a elektronických zvětšovacích (kamerové lupy) kompenzačních pomůcek. Nositelé patogenních mutací by se měli přísně vyvarovat kouření (i pasivnímu), nadměrné konzumaci alkoholu a výše zmíněné medikaci s mitochondriální toxicitou [23,37]. U pacientů s extraokulární manifestací je nutná dispenzarizace na specializovaných kardiologických a neurologických pracovištích.
Do nedávné doby byl jedinou terapeutickou možností koenzym Q10, který prostřednictvím sukcinátdehydrogenázy (mitochondriální komplex II) obchází nefunkční komplex I, a zvyšuje tak produkci ATP systémem oxidativní fosforylace. Tato látka je však silně lipofilní a při perorálním podávání je jeho průnik do mitochondrie velmi diskutabilní [98]. Jeho účinnost nebyla nikdy prokázána v klinické studii.
V posledních letech bylo provedeno několik studií testujících nové léčebné přípravky s předpokládaným pozitivním vlivem na stabilizaci a návrat zrakových funkcí. Slibná jsou především analoga ubichinonu s krátkým postranním řetězcem, jakými jsou idebenone a α-tokotrienolchinon (EPI-743), která nahrazují funkci dysfunkčního komplexu I a snižují produkci ROS [99]. Idebenone navíc prokazatelně reaktivuje inaktivní, ale stále životaschopné RGB, čímž umožňuje návrat zrakových funkcí i více než rok po vzniku onemocnění [100].
V randomizované dvojitě placebem zaslepené studii RHODOS (Rescue of Hereditary Optic Disease Outpatient Study), ve které bylo zhodnoceno 82 pacientů s mutací m.11778G>A, došlo při podávání perorálního idebenone per os v dávce 900 mg/den k prokazatelnému zlepšení nejlepší korigované zrakové ostrosti, barvocitu a návratu zrakových funkcí [101]. Po 24 týdnech se zlepšila zraková ostrost především u podskupiny pacientů s dominantním postižením jednoho oka (n = 30). Ve 24. týdnu studie vedla terapie idebenonem ke zlepšení logMAR o ≥ 0,2 u 55 % pacientů, oproti 10 % pacientů v placebo skupině. Žádnému z pacientů léčených idebenonem se zrakové funkce nezhoršily do pásma praktické slepoty [101]. V současnosti je idebenone jedinou oficiálně schválenou léčbou LHON v České republice. Léčba přípravkem EPI-743 je zatím ve fázi klinických zkoušek (IIa) [102]. Pilotní studie s 5 pacienty s LHON užívajícícími EPI-743 prokázala stabilizaci onemocnění a návrat zrakových funkcí ve čtyřech případech, přičemž u dvou jedinců byl návrat zrakových funkcí kompletní [102].
V září 2015 obdržela firma GenSight Biologics povolení zahájit fázi III. klinických zkoušek genové terapie GS010 pro pacienty s LHON nesoucími mutaci m.11778G>A [103]. Terapie je založena na jednorázové intravitreální injekci rekombinantního adeno-asociovaného viru 2 nesoucího gen kódující ND4 podjednotku komplexu I (rAAV2-ND4). První studie Wan et al ukázala významné zlepšení zrakové ostrosti a nálezu na perimetru na očích 9 pacientů, avšak tloušťka RNFL a prodloužená odezva na VEP se nezměnila [104]. K vyhodnocení účinnosti této nadějné léčby je třeba vyčkat na výsledky dlouhodobé, multicentrické, randomizované studie.
Na experimentální úrovni je studována celá řada dalších látek. Jednu skupinu tvoří aktivátory mitochondriální biogeneze (resveratrol, bezafibrát, rosiglitazon a 5-aminoimidazol-4-karboxamid ribonukleosid neboli AICAR), které by mohly zvyšovat kompenzační potenciál symptomatických pacientů [105]. Nadějně se jeví i ovlivnění estrogenových receptorů pomocí fytoestrogenů. Stimulace ER-β receptorů na myších modelech vedla ke snížení produkce ROS, a tudíž i apoptózy, a podpoře mitochondriální biogeneze [106]. Obdobně měl na myších modelech antiapoptotické a antioxidativní účinky i α2-agonista brimonidin [107]. Přestože u pacientů s izolovaným LHON neměl prokazatelné účinky na zlepšení zrakových funkcí, u pacientů s LHON a současně diagnostikovaným glaukomem, který zhoršuje stázu axoplazmy, se léčba brimonidinem jeví jako racionální [98,108,109].
Centralizace péče o pacienty s LHON a jinými neuropatiemi optiku
Nové možnosti terapie LHON podtrhují nutnost časné diagnózy. Adekvátní léčba bývá bohužel často zahájena pozdě, a nedosahuje tak požadovaných výsledků. Z výše uvedených důvodů vzniklo ve VFN v Praze specializované Centrum pro pacienty s mitochondriálními neuropatiemi optiku za účelem optimalizace diagnostických a terapeutických algoritmů a studia rizikových faktorů rozvoje mitochondriálních onemocnění postihujících také zrakový nerv. V roce 2016 byla Oční klinika 1. LF UK a VFN v Praze zapojena do Evropské referenční sítě pro diagnostiku vzácných onemocnění oka (ERN-EYE) a právě Centrum pro pacienty s mitochondriálními neuropatiemi optiku je i v mezinárodním kontextu její významnou součástí (obr. 6). Hlavním cílem této evropské iniciativy je posílení postavení pacientů a podpora vysoce kvalitní, komplexní a nákladově efektivní zdravotní péče pro pacienty s vzácnými chorobami, a to zejména prostřednictvím úzké spolupráce specializovaných zdravotnických zařízení v rámci celé Evropské unie.

V průběhu dvou let bylo v Centru pro pacienty s mitochondriálními neuropatiemi optiku podrobně vyšetřeno celkem 54 jedinců (20 symptomatických) s LHON, 5 symptomatických pacientů s DOA [110] a 10 pacientů s neuropatií optiku se zatím neobjasněnou příčinou. U 2 pacientů s neznámou příčinou atrofie se podařilo odhalit kauzální mutace v podjednotkách komplexu I [39]. Díky včasné diagnóze stanovené v našem centru bylo u 9 pacientů s LHON možno zahájit terapii idebenonem v období od 4 týdnů do 9 měsíců po vzniku ztráty zraku. Dva pacienty se podařilo zařadit do fáze III. klinických zkoušek genové terapie v Nemocnici Mnichov-Schwabing (RESCUE Study, GenSight Biologics, Paris; číslo studie: GS-LHON-CLIN-03A) a jsou tak vůbec prvními pacienty léčenými genovou terapií v České republice.
Závěr
LHON je maternálně dědičné onemocnění projevující se rychlou, nebolestivou ztrátou zraku. Zcela nedávno bylo prokázáno, že včasné zahájení terapie idebenonem může vést k významnému zlepšení zrakových funkcí u pacientů s manifestací tohoto onemocnění. Vzhledem k tomu, že LHON bývá často nesprávně vyhodnocen a následně i léčen jako RS, od které je v akutní fázi prakticky neodlišitelný, měl by být zavzat do diferenciálně diagnostické rozvahy u každé progresivně se rozvíjející neuropatie optiku.
Práce byla podpořena grantem AZV 16-32341A a výzkumnými záměry SVV 260367, VFN-RVO 64165, UNCE 204011 a PROGRES-Q26/ LF1.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
doc. MUDr. Petra Lišková, MD, Ph.D.
Ústav dědičných metabolických poruch
1. LF UK a VFN v Praze
Ke Karlovu 455/2
128 08 Praha 2
e-mail: Petra.Liskova@lf1.cuni.cz
Přijato k recenzi: 18. 3. 2017
Přijato do tisku: 1. 8. 2017
Sources
1. Schaefer AM, Taylor RW, Turnbull DM, et al. The epidemiology of mitochondrial disorders – past, present and future. Biochim Biophys Acta 2004;1659(2 – 3):115 – 20.
2. Leber T. About hereditary and congenital optic neuropathy. Albrecht Von Grafes Arch Ophthalmol 1871;17 : 249 – 91.
3. Wallace DC. A new manifestation of Leber‘s disease and a new explanation for the agency responsible for its unusual pattern of inheritance. Brain 1970;93(1):121 – 32.
4. Wallace DC, Singh G, Lott MT, et al. Mitochondrial DNA mutation associated with Leber‘s hereditary optic neuropathy. Science 1988;242(4884):1427 – 30.
5. Carelli V, Ross-Cisneros FN, Sadun AA. Mitochondrial dysfunction as a cause of optic neuropathies. Prog Retin Eye Res 2004;23(1):53 – 89.
6. Yu-Wai-Man P, Turnbull DM, Chinnery PF. Leber hereditary optic neuropathy. J Med Genet 2002;39(3):162 – 9.
7. Yu-Wai-Man P, Griffiths PG, Brown DT, et al. The epidemiology of Leber hereditary optic neuropathy in the North East of England. Am J Hum Genet 2003;72(2):333 – 9.
8. Gorman GS, Schaefer AM, Ng Y, et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann Neurol 2015;77(5): 753 – 9. doi: 10.1002/ ana.24362.
9. Spruijt L, Kolbach DN, de Coo RF, et al. Influence of mutation type on clinical expression of Leber hereditary optic neuropathy. Am J Ophthalmol 2006;141(4):676 – 82.
10. Puomila A, Hamalainen P, Kivioja S, et al. Epidemiology and penetrance of Leber hereditary optic neuropathy in Finland. Eur J Hum Genet 2007;15(10):1079 – 89.
11. Yu-Wai-Man P, Chinnery PF. Leber Hereditary Optic Neuropathy. In: Pagon RA, Adam MP, Ardinger HH, et al. eds. Seattle: Seattle University of Washington 1993.
12. Nikoskelainen EK, Huoponen K, Juvonen V, et al. Ophthalmologic findings in Leber hereditary optic neuropathy, with special reference to mtDNA mutations. Ophthalmology 1996;103(3):504 – 14.
13. Riordan-Eva P, Sanders MD, Govan GG, et al. The clinical features of Leber‘s hereditary optic neuropathy defined by the presence of a pathogenic mitochondrial DNA mutation. Brain 1995; 118(Pt 2):319 – 37.
14. Dimitriadis K, Leonhardt M, Yu-Wai-Man P, et al. Leber‘s hereditary optic neuropathy with late disease onset: clinical and molecular characteristics of 20 patients. Orphanet J Rare Dis 2014;9 : 158. doi: 10.1186/ s13023-014-0158-9.
15. Barboni P, Carbonelli M, Savini G, et al. Natural history of Leber‘s hereditary optic neuropathy: longitudinal analysis of the retinal nerve fiber layer by optical coherence tomography. Ophthalmology 2010;117(3):623 – 27. doi: 10.1016/ j.ophtha.2009.07.026.
16. Wakakura M, Yokoe J. Evidence for preserved direct pupillary light response in Leber‘s hereditary optic neuropathy. Br J Ophthalmol 1995;79(5):442 – 6.
17. Kawasaki A, Herbst K, Sander B, et al. Selective wavelength pupillometry in Leber hereditary optic neuropathy. Clin Exp Ophthalmol 2010;38(3):322 – 4. doi: 10.1111/ j.1442-9071.2010.02212.x.
18. Ziccardi L, Sadun F, De Negri AM, et al. Retinal function and neural conduction along the visual pathways in affected and unaffected carriers with Leber‘s hereditary optic neuropathy. Invest Ophthalmol Vis Sci 2013;54(10):6893 – 901. doi: 10.1167/ iovs.13-12894.
19. Nikoskelainen E, Sogg RL, Rosenthal AR, et al. The early phase in Leber hereditary optic atrophy. Arch Ophthalmol 1977;95(6):969 – 78.
20. Harding AE, Sweeney MG, Govan GG, et al. Pedigree analysis in Leber hereditary optic neuropathy families with a pathogenic mtDNA mutation. Am J Hum Genet 1995;57(1):77 – 86.
21. Barboni P, Savini G, Valentino ML, et al. Retinal nerve fiber layer evaluation by optical coherence tomography in Leber‘s hereditary optic neuropathy. Ophthalmology 2005;112(1):120 – 6.
22. Balducci N, Savini G, Cascavilla ML, et al. Macular nerve fibre and ganglion cell layer changes in acute Leber‘s hereditary optic neuropathy. Br J Ophthalmol 2016;100(9):1232 – 7. doi: 10.1136/ bjophthalmol-2015-307326.
23. Kirkman MA, Yu-Wai-Man P, Korsten A, et al. Gene-environment interactions in Leber hereditary optic neuropathy. Brain 2009;132(Pt 9):2317 – 26. doi: 10.1093/ brain/ awp158.
24. Barboni P, Savini G, Valentino ML, et al. Leber‘s hereditary optic neuropathy with childhood onset. Invest Ophthalmol Vis Sci 2006;47(12):5303 – 9. doi: 10.1167/ iovs.06-0520.
25. Ramos Cdo V, Bellusci C, Savini G, et al. Association of optic disc size with development and prognosis of Leber‘s hereditary optic neuropathy. Invest Ophthalmol Vis Sci 2009;50(4):1666–74. doi: 10.1167/ iovs.08-2695.
26. Riordan-Eva P, Harding AE. Leber‘s hereditary optic neuropathy: the clinical relevance of different mitochondrial DNA mutations. J Med Genet 1995;32(2):81 – 7.
27. Johns DR, Heher KL, Miller NR, et al. Leber‘s hereditary optic neuropathy. Clinical manifestations of the 14484 mutation. Arch Ophthalmol 1993;111(4):495 – 8.
28. Macmillan C, Kirkham T, Fu K, et al. Pedigree analysis of French Canadian families with T14484C Leber‘s hereditary optic neuropathy. Neurology 1998;50(2):417 – 22.
29. Johns DR, Neufeld MJ, Park RD. An ND-6 mitochondrial DNA mutation associated with Leber hereditary optic neuropathy. Biochem Biophys Res Commun 1992;187(3):1551 – 7.
30. Lam BL, Feuer WJ, Schiffman JC, et al. Trial end points and natural history in patients with G11778A Leber hereditary optic neuropathy : preparation for gene therapy clinical trial. JAMA Ophthalmology 2014;132(4):428 – 36. doi: 10.1001/ jamaophthalmol.2013.7971.
31. Newman NJ, Lott MT, Wallace DC. The clinical characteristics of pedigrees of Leber‘s hereditary optic neuropathy with the 11778 mutation. Am J Ophthalmol 1991;111(6):750 – 62.
32. Sadun AA, Salomao SR, Berezovsky A, et al. Subclinical carriers and conversions in Leber hereditary optic neuropathy: a prospective psychophysical study. Trans Am Ophthalmol Soc 2006;104 : 751 – 61.
33. Barboni P, Savini G, Valentino ML, et al. Retinal nerve fiber layer evaluation by optical coherence tomography in unaffected carriers with Leber‘s hereditary optic neuropathy mutations. Ophthalmology 2005;112(1):127 – 31.
34. Salomao SR, Berezovsky A, Andrade RE, et al. Visual electrophysiologic findings in patients from an extensive Brazilian family with Leber‘s hereditary optic neuropathy. Doc Ophthalmol 2004;108(2):147 – 55.
35. Sacai PY, Salomao SR, Carelli V, et al. Visual evokedpotentials findings in non-affected subjects from a large Brazilian pedigree of 11778 Leber‘s hereditary optic neuropathy. Doc Ophthalmol 2010;121(2):147 – 54. doi: 10.1007/ s10633-010-9241-2.
36. Livingstone IR, Mastaglia FL, Howe JW, et al. Leber‘s optic neuropathy: clinical and visual evoked response studies in asymptomatic and symptomatic members of a 4-generation family. Br J Ophthalmol 1980;64(10):751 – 7.
37. Yu-Wai-Man P, Griffiths PG, Chinnery PF. Mitochondrial optic neuropathies - disease mechanisms and therapeutic strategies. Prog Retin Eye Res 2011;30(2):81 – 114. doi: 10.1016/ j.preteyeres.2010.11.002.
38. Blakely EL, de Silva R, King A, et al. LHON/ MELAS overlap syndrome associated with a mitochondrial MTND1 gene mutation. Eur J Hum Genet 2005;13(5):623 – 7.
39. Kolarova H, Liskova P, Tesarova M et al. Unique presentation of LHON/ MELAS overlap syndrome caused by m.13046T>C in MTND5. Ophthalmic Genet 2016;37(4):419 – 23.
40. Howell N, Kubacka I, Xu M, et al. Leber hereditary optic neuropathy: involvement of the mitochondrial ND1 gene and evidence for an intragenic suppressor mutation. Am J Hum Genet 1991;48(5):935 – 42.
41. De Vries DD, Went LN, Bruyn GW, et al. Genetic and biochemical impairment of mitochondrial complex I activity in a family with Leber hereditary optic neuropathy and hereditary spastic dystonia. Am J Hum Genet 1996;58(4):703 – 11.
42. Jun AS, Brown MD, Wallace DC. A mitochondrial DNA mutation at nucleotide pair 14459 of the NADH dehydrogenase subunit 6 gene associated with maternally inherited Leber hereditary optic neuropathy and dystonia. Proc Natl Acad Sci USA 1994;91(13):6206 – 10.
43. Gropman A, Chen TJ, Perng CL, et al. Variable clinical manifestation of homoplasmic G14459A mitochondrial DNA mutation. Am J Med Genet A 2004;124A(4):377 – 82.
44. Tarnopolsky MA, Baker SK, Myint T, et al. Clinical variability in maternally inherited leber hereditary optic neuropathy with the G14459A mutation. Am J Med Genet A 2004; 124A(4):372 – 6. doi: 10.1002/ ajmg.a.20449.
45. Newman NJ. Leber‘s hereditary optic neuropathy. New genetic considerations. Arch Neurol 1993;50(5):540 – 8.
46. Harding AE, Sweeney MG, Miller DH, et al. Occurrence of a multiple sclerosis-like illness in women who have a Leber‘s hereditary optic neuropathy mitochondrial DNA mutation. Brain 1992;115(Pt 4):979 – 89.
47. Dotti MT, Caputo N, Signorini E, et al. Magnetic resonance imaging findings in Leber‘s hereditary optic neuropathy. Eur Neurol 1992;32(1):17 – 9.
48. Mashima Y, Oshitari K, Imamura Y, et al. Orbital high resolution magnetic resonance imaging with fast spin echo in the acute stage of Leber‘s hereditary optic neuropathy. J Neurol Neurosurg Psychiatry 1998;64(1):124 – 7.
49. Kellar-Wood H, Robertson N, Govan GG, et al. Leber‘s hereditary optic neuropathy mitochondrial DNA mutations in multiple sclerosis. Ann Neurol 1994;36(1):109 – 12.
50. Jansen PH, van der Knaap MS, de Coo IF. Leber‘s hereditary optic neuropathy with the 11,778 mtDNA mutation and white matter disease resembling multiple sclerosis: clinical, MRI and MRS findings. J Neurol Sci 1996;135(2):176 – 80.
51. Vanopdenbosch L, Dubois B, D‘Hooghe MB, et al. Mitochondrial mutations of Leber‘s hereditary optic neuropathy: a risk factor for multiple sclerosis. J Neurol 2000;247(7):535 – 43.
52. Pfeffer G, Burke A, Yu-Wai-Man P, et al. Clinical features of MS associated with Leber hereditary optic neuropathy mtDNA mutations. Neurology 2013;81(24):2073 – 81. doi: 10.1212/ 01.wnl.0000437308.22603.43.
53. Matthews L, Enzinger C, Fazekas F, et al. MRI in Leber‘s hereditary optic neuropathy: the relationship to multiple sclerosis. J Neurol Neurosurg Psychiatry 2015;86(5):537 – 42. doi: 10.1136/ jnnp-2014-308186.
54. Palace J. Multiple sclerosis associated with Leber‘s Hereditary Optic Neuropathy. J Neurol Sci 2009;286(1-2):24 – 7. doi: 10.1016/ j.jns.2009.09.009.
55. Bristow EA, Griffiths PG, Andrews RM, et al. The distribution of mitochondrial activity in relation to optic nerve structure. Arch Ophthalmol 2002;120(6):791 – 6.
56. Howell N. Leber hereditary optic neuropathy: respiratory chain dysfunction and degeneration of the optic nerve. Vision Res 1998;38(10):1495 – 504.
57. Lowry OH, Roberts NR, Lewis C. The quantitative histochemistry of the retina. J Biol Chem 1956;220(2):879 – 92.
58. Kageyama GH, Wong-Riley MT. The histochemical localization of cytochrome oxidase in the retina and lateral geniculate nucleus of the ferret, cat, and monkey, with particular reference to retinal mosaics and ON/ OFF-center visual channels. J Neurosci 1984;4(10):2445 – 59.
59. Klivenyi P, Karg E, Rozsa C, et al. Alpha-Tocopherol/ lipid ratio in blood is decreased in patients with Leber‘s hereditary optic neuropathy and asymptomatic carriers of the 11778 mtDNA mutation. J Neurol Neurosurg Psychiatry 2001;70(3):359 – 62.
60. Alexander C, Votruba M, Pesch UE, et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet 2000;26(2):211 – 5.
61. Delettre C, Lenaers G, Griffoin JM, et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet 2000;26(2):207 – 10.
62. Giordano C, Iommarini L, Giordano L, et al. Efficient mitochondrial biogenesis drives incomplete penetrance in Leber‘s hereditary optic neuropathy. Brain 2014;137(Pt 2):335 – 53. doi: 10.1093/ brain/ awt343.
63. Sadun AA, La Morgia C, Carelli V. Mitochondrial optic neuropathies: our travels from bench to bedside and back again. Clin Exp Ophthalmol 2013;41(7):702 – 12. doi: 10.1111/ ceo.12086.
64. Vidrova V, Tesarova M, Trefilova E, et al. Mitochondrial DNA haplogroups in the Czech population compared to other European countries. Hum Biol 2008;80(6):669–74. doi: 10.3378/ 1534-6617-80.6.669.
65. Wallace DC, Brown MD, Lott MT. Mitochondrial DNA variation in human evolution and disease. Gene 1999;238(1):211 – 30.
66. Howell N, Kubacka I, Halvorson S, et al. Leber‘s hereditary optic neuropathy: the etiological role of a mutation in the mitochondrial cytochrome b gene. Genetics 1993;133(1):133 – 6.
67. Brown MD, Torroni A, Reckord CL, et al. Phylogenetic analysis of Leber‘s hereditary optic neuropathy mitochondrial DNA‘s indicates multiple independent occurrences of the common mutations. Hum Mutat 1995;6(4):311–25.
68. Vergani L, Martinuzzi A, Carelli V, et al. MtDNA mutations associated with Leber‘s hereditary optic neuropathy: studies on cytoplasmic hybrid (cybrid) cells. Biochem Biophys Res Commun 1995;210(3):880 – 8.
69. Brown MD, Sun F, Wallace DC. Clustering of Caucasian Leber hereditary optic neuropathy patients containing the 11778 or 14484 mutations on an mtDNA lineage. Am J Hum Genet 1997;60(2):381 – 7.
70. Torroni A, Petrozzi M, D‘Urbano L, et al. Haplotype and phylogenetic analyses suggest that one European-specific mtDNA background plays a role in the expression of Leber hereditary optic neuropathy by increasing the penetrance of the primary mutations 11778 and 14484. Am J Hum Genet 1997;60(5):1107 – 21.
71. Torroni A, Wallace DC. Mitochondrial DNA variation in human populations and implications for detection of mitochondrial DNA mutations of pathological significance. J Bioenerg Biomembr 1994;26(3):261 – 71.
72. Smith KH, Johns DR, Heher KL, et al. Heteroplasmy in Leber‘s hereditary optic neuropathy. Arch Ophthalmol 1993;111(11):1486 – 90.
73. Chinnery PF, Andrews RM, Turnbull DM, et al. Leber hereditary optic neuropathy: Does heteroplasmy influence the inheritance and expression of the G11778A mitochondrial DNA mutation? Am J Med Genet 2001;98(3):235 – 43.
74. Black GC, Morten K, Laborde A, et al. Leber‘s hereditary optic neuropathy: heteroplasmy is likely to be significant in the expression of LHON in families with the 3460 ND1 mutation. Br J Ophthalmol 1996;80(10):915 – 7.
75. Jacobi FK, Leo-Kottler B, Mittelviefhaus K, et al. Segregation patterns and heteroplasmy prevalence in Leber‘s hereditary optic neuropathy. Invest Ophthalmol Vis Sci 2001;42(6):1208 – 14.
76. Howell N, Xu M, Halvorson S, et al. A heteroplasmic LHON family: tissue distribution and transmission of the 11778 mutation. Am J Hum Genet 1994;55(1):203 – 6.
77. Weber K, Wilson JN, Taylor L, et al. A new mtDNA mutation showing accumulation with time and restriction to skeletal muscle. Am J Hum Genet 1997;60(2):373 – 80.
78. Howell N, Ghosh SS, Fahy E, et al. Longitudinal analysis of the segregation of mtDNA mutations in heteroplasmic individuals. J Neurol Sci 2000;172(1):1 – 6.
79. Bu XD, Rotter JI. X chromosome-linked and mitochondrial gene control of Leber hereditary optic neuropathy: evidence from segregation analysis for dependence on X chromosome inactivation. Proc Natl Acad Sci USA 1991;88(18):8198 – 202.
80. Nakamura M, Fujiwara Y, Yamamoto M. The two locus control of Leber hereditary optic neuropathy and a high penetrance in Japanese pedigrees. Hum Genet 1993;91(4):339 – 41.
81. Carvalho MR, Muller B, Rotzer E, et al. Leber‘s hereditary optic neuroretinopathy and the X-chromosomal susceptibility factor: no linkage to DXs7. Hum Hered 1992;42(5):316 – 20.
82. Chen JD, Cox I, Denton MJ. Preliminary exclusion of an X-linked gene in Leber optic atrophy by linkage analysis. Hum Genet 1989;82(3):203 – 7.
83. Sweeney MG, Davis MB, Lashwood A, et al. Evidence against an X-linked locus close to DXS7 determining visual loss susceptibility in British and Italian families with Leber hereditary optic neuropathy. Am J Hum Genet 1992;51(4):741 – 8.
84. Handoko HY, Wirapati PJ, Sudoyo HA, et al. Meiotic breakpoint mapping of a proposed X linked visual loss susceptibility locus in Leber‘s hereditary optic neuropathy. J Med Genet 1998;35(8):668 – 71.
85. Giordano C, Montopoli M, Perli E, et al. Oestrogens ameliorate mitochondrial dysfunction in Leber‘s hereditary optic neuropathy. Brain 2011;134(Pt 1):220 – 34. doi: 10.1093/ brain/ awq276.
86. Pott JW, Wong KH. Leber‘s hereditary optic neuropathy and vitamin B12 deficiency. Graefes Arch Clin Exp Ophthalmol 2006;244(10):1357 – 9.
87. Jalil A, Usmani HA, Khan MI, et al. Bilateral paediatric optic neuropathy precipitated by vitamin B12 deficiency and a novel mitochondrial DNA mutation. Int Ophthalmol 2013;33(6):687 – 90. doi: 10.1007/ s10792-013-9773-z.
88. Saini M, Khurana D. Chronic relapsing inflammatory optic neuropathy. Ann Indian Acad Neurol 2010;13(1):61 – 3. doi: 10.4103/ 0972-2327.61280.
89. Scolding N. The differential diagnosis of multiple sclerosis. J Neurol Neurosurg Psychiatry 2001;71(Suppl 2):ii9 – 15.
90. Kidd D, Burton B, Plant GT, et al. Chronic relapsing inflammatory optic neuropathy (CRION). Brain 2003;126(Pt 2):276 – 84.
91. Germann CA, Baumann MR, Hamzavi S. Ophthalmic diagnoses in the ED: optic neuritis. Am J Emerg Med 2007;25(7):834 – 7.
92. Igarashi Y, Oyachi H, Nakamura Y, et al. Neuromyelitis optica. Ophthalmologica 1994;208(4):226 – 9.
93. Arnold TW, Myers GJ. Neuromyelitis optica (Devic syndrome) in a 12-year-old male with complete recovery following steroids. Pediatr Neurol 1987;3(5):313 – 5.
94. Hinson SR, Pittock SJ, Lucchinetti CF, et al. Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology 2007;69(24):2221 – 31.
95. Saadoun S, Waters P, Bell BA, et al. Intra-cerebralinjection of neuromyelitis optica immunoglobulin Gand human complement produces neuromyelitis optica lesions in mice. Brain 2010;133(Pt 2):349 – 61. doi: 10.1093/ brain/ awp309.
96. Waters P, Vincent A. Detection of anti-aquaporin-4 antibodies in neuromyelitis optica: current status of the assays. Int MS J 2008;15(3):99 – 105.
97. Votruba M, Moore AT, Bhattacharya SS. Clinical features, molecular genetics, and pathophysiology of dominant optic atrophy. J Med Genet 1998;35(10):793 – 800.
98. Meyerson C, Van Stavern G, McClelland C. Leber hereditary optic neuropathy: current perspectives. Clin Ophthalmol 2015;9 : 1165 – 76. doi: 10.2147/ opth.s62021.
99. Geromel V, Darin N, Chretien D, et al. Coenzyme Q(10) and idebenone in the therapy of respiratory chain diseases: rationale and comparative benefits. Mol Genet Metab 2002;77(1 – 2):21 – 30.
100. Lyseng-Williamson KA. Idebenone: A Review in Leber‘s Hereditary Optic Neuropathy. Drugs 2016;76(7):805 – 13. doi: 10.1007/ s40265-016-0574-3.
101. Klopstock T, Yu-Wai-Man P, Dimitriadis K, et al. A randomized placebo-controlled trial of idebenone in Leber‘s hereditary optic neuropathy. Brain 2011;134(Pt 9):2677 – 86. doi: 10.1093/ brain/ awr170.
102. Sadun AA, Chicani CF, Ross-Cisneros FN, et al. Effect of EPI-743 on the clinical course of the mitochondrial disease Leber hereditary optic neuropathy. Arch Neurol 2012;69(3):331 – 8. doi: 10.1001/ archneurol.2011.2972.
103. GenSight. GenSight Biologics Receives IND Acceptance from FDA to Enter Phase III with GS010. Paris, France; 2015.
104. Wan X, Pei H, Zhao MJ, et al. Efficacy and Safety of rAAV2-ND4 Treatment for Leber‘s Hereditary Optic Neuropathy. Sci Rep 2016;6 : 21587. doi: 10.1038/ srep21587.
105. La Morgia C, Carbonelli M, Barboni P, et al. Medical management of hereditary optic neuropathies. Front Neurol 2014;5 : 141. doi: 10.3389/ fneur.2014.00141.
106. Pisano A, Preziuso C, Iommarini L, et al. Targeting estrogen receptor beta as preventive therapeutic strategy for Leber‘s hereditary optic neuropathy. Hum Mol Genet 2015;24(24):6921 – 31. doi: 10.1093/ hmg/ ddv396.
107. Saylor M, McLoon LK, Harrison AR, et al. Experimental and clinical evidence for brimonidine as an optic nerve and retinal neuroprotective agent: an evidence-based review. Arch Ophthalmol 2009;127(4):402 – 6. doi: 10.1001/ archophthalmol.2009.9.
108. Newman NJ, Biousse V, David R, et al. Prophylaxis for second eye involvement in leber hereditary optic neuropathy: an open-labeled, nonrandomized multicenter trial of topical brimonidine purite. Am J Ophthalmol 2005;140(3):407 – 15. doi: 10.1016/ j.ajo.2005.03.058.
109. Thouin A, Griffiths PG, Hudson G, et al. Raised intraocular pressure as a potential risk factor for visual loss in Leber Hereditary Optic Neuropathy. PLoS One 2013;8(5):e63446. doi: 10.1371/ journal.pone.0063446.
110. Liskova P, Tesarova M, Dudakova L, et al. OPA1 analysis in an international series of probands with bilateral optic atrophy. Acta Ophthalmol 2017;95(4):363 – 9. doi: 10.1111/ aos.13285.
111. Moraes CT, DiMauro S, Zeviani M, et al. Mitochondrial DNA deletions in progressive external ophthalmoplegia and Kearns-Sayre syndrome. N Engl J Med 1989;320(20):1293 – 9.
112. Nikoskelainen EK. Clinical picture of LHON. ClinNeurosci 1994;2 : 115 – 20.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2017 Issue 5
Most read in this issue
- Essential Tremor – Is There a New Nosological Concept?
- Leber Hereditary Optic Neuropathy
- Statin-induced Necrotizing Autoimmune Myopathy
- Invasive Methods in the Treatment of Advanced Parkinson’s Disease