
Genetické a epigenetické faktory podmiňující vznik a prognózu mozkových gliomů – souhrn současných poznatků
Genetic and Epigenetic Factors Affecting Development and Prognosis of Brain Gliomas – a Review of Current Knowledge
Gliomas represent a heterogenous group of primary brain tumors that can significantly vary in prognosis and treatment. The WHO Brain Tumors classification is based on morphological criteria that seem inadequate as they do not reflect new molecular markers. These new markers affect prognosis, overall survival and treatment more than histological diagnosis. IDH1/2 mutation and 1p/19q codeletion are the most important molecular markers predicting patient prognosis in lower grade gliomas and secondary glioblastomas. IDH1/2 mutation is a marker of better prognosis than wild type IDH. Similarly, ATRX plays an important role in anaplastic astrocytomas and loss of ATRX expression positively affects treatment response in these tumors. Clinical studies suggest that 1p/19q codeletion is the most relevant prognostic marker in oligodendroglial tumors, probably because of tumor chemosensitivity. Besides 1p/19q codeletion, TERT promoter mutation is another important factor in overall survival. Patients with gliomas carrying combined IDH1/2 mutation and 1p/19q codeletion have the best prognosis compared to those with wild type IDH. Glioblastomas are highly malignant glial tumors. Despite the progress in understanding of tumor formation and development, they are still difficult to treat. Nevertheless, MGMT promoter methylation in GBM is the most important predictor of good treatment response. A specific EGFRvIII mutation is another potential treatment target. Recently, single nucleotide polymorphisms (SNP) were found as another, in this case inherited, factor that can have an impact on a patient´s prognosis and treatment response. In particular, rs55705857 in anaplastic oligodendroglioma G allele carriers have much better prognosis in comparison to those who carry A allele. These new findings confirm that the prognosis is affected by multiple factors, including inherited predisposition.
Key words:
glioma – diffuse astrocytoma – anaplastic astrocytoma – oligodendroglioma – anaplastic oligodendroglioma – glioblastoma – molecular markers – single nucleotide polymorphism
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Authors:
F. Kramář 1; M. Minárik 2,3; B. Belšánová 2; T. Hálková 2; O. Bradáč 1; D. Netuka 1; V. Beneš 1
Authors‘ workplace:
Neurochirurgická a neuroonkologická klinika 1. LF UK a ÚVN – VFN Praha
1; Centrum aplikované genomiky solidních nádorů (CEGES), Genomac výzkumný ústav, s. r. o., Praha
2; Katedra analytické chemie, PřF UK v Praze
3
Published in:
Cesk Slov Neurol N 2016; 79/112(4): 400-405
Category:
Review Article
doi:
https://doi.org/10.14735/amcsnn2016400
Overview
Gliomy představují heterogenní skupinu primárních mozkových nádorů, jejichž prognóza a léčba se mohou významně lišit. WHO klasifikace je založena na morfologických kritériích, což se ukazuje jako nedostatečné. Nicméně v poslední době se do popředí stále více dostávají molekulárně genetické markery, které ovlivňují prognózu, terapeutickou účinnost a celkové přežití daleko více než samotná histologická kritéria. Především mutace IDH1/ 2 a kodelece 1p/ 19q představují v současné době nejvýznamnější molekulární znaky pro stanovení prognózy u gliomů nižších stupňů. U anaplastických astrocytomů hraje významnou roli v prognóze exprese ATRX, u oligodendrogliomů je to vedle kodelece 1p/ 19q mutace promotoru TERT. Glioblastomy i nadále představují terapeuticky obtížně ovlivnitelné onemocnění, hlavním markerem prognózy zůstává metylace promotoru MGMT a případně mutace EGFRvIII. V nedávné době byla odhalena i role jednobodových polymorfizmů – SNP. Jedná se o záměny bazí, které se vyskytují i ve zdravé populaci. V současné době známe více takovýchto polymorfních úseků, u kterých byl vysledován vztah k predispozici nebo prognóze pacientů s gliomy.
Klíčová slova:
gliom – difuzní astrocytom – anaplastický astrocytom – oligodendrogliom – anaplastický oligodendrogliom – glioblastom – molekulární markery – jednonukleotidový polymorfizmus
Úvod
Gliomy představují heterogenní skupinu primárních mozkových nádorů, jejichž prognóza a léčba se mohou významně lišit. Incidence gliálních nádorů dosahuje cca 4,5– 5 případů na 100 000 obyvatel/ rok. WHO klasifikace je založena na morfologických kritériích, což se ukazuje jako nedostatečné pro prognózu i léčbu. Nicméně v poslední době se do popředí stále více dostávají molekulárně genetické markery, které naopak ovlivňují prognózu, terapeutickou účinnost a celkové přežití daleko více než samotná histologická kritéria. V některých případech lze říci, že histologická analýza má již pouze informativní charakter a naopak molekulární markery zásadně ovlivňují prognózu pacientů. Navíc je souladu histologické analýzy s molekulárními znaky dosaženo pouze v asi 60 % případů [1]. Prozatím tato znalost pouze v malé míře ovlivňuje nasazení účinné léčby, ale s tím, jak se naše poznatky o patogenezi těchto onemocnění rozšiřují, zvyšují se i možnosti cílené molekulárně genetické diagnostiky a z toho vyplývající léčby [2,3].
IDH1/ 2
Dlouhou dobu se předpokládalo, že jediným možným mechanizmem vzniku mozkových nádorů jsou genetické alterace (mutace/ delece) onkogenů nebo naopak tumor-supresorových genů, které nakonec vedou ke vzniku nádoru. Tuto hypotézu podporuje celá řada důkazů a známe nádory, kde tento mechanizmus vzniku je velice častý (např. primární glioblastom). Až do roku 2009 jsme ani jiný mechanizmus vzniku neznali. Teprve práce The Cancer Genome Atlas Research Network, která se zabývala glioblastomy (GBM), odhalila, že u části GBM se vyskytuje mutace isocitrátdehydrogenázy 1 (IDH1) [4], metabolického enzymu, který do té doby s karcinogenezí nebyl spojován. To vedlo k dalšímu zkoumání, zda má tato mutace nějaký význam pro tumorigenezi gliomů. A skutečně bylo prokázáno, že mutace IDH1 se vyskytuje ve vysokém procentu gliomů nižších stupnů (až 85 %), a to jak astrocytárních, tak oligodendrogliálních nádorů. V lidském těle se nachází celkem tři izoformy isocitrátdehydrogenázy – IDH1– 3. Dalším výzkumem bylo prokázáno, že podobná mutace může ovlivňovat a měnit také enzymatickou aktivitu IDH2, ale nikdy IDH3. Vyplývá to ze struktury jednotlivých enzymů, IDH1 a 2 jsou homodimery, zatímco IDH3 je heterotetramer. Ve všech případech mutace IDH1/ 2 se jedná o missense mutaci postihující kodon pro arginin, R132H v případě IDH1 nebo ekvivalentně R172H u IDH2 (mutace se nachází v aktivním místě enzymu, arginin je v proteinovém řetězci nahrazen histidinem). Mutace vede ke změně struktury a enzymatické aktivity tohoto enzymu, který v rámci citrátového cyklu katalyzuje konverzi isocitrátu na α-ketoglutarát. Tato změněná aktivita enzymu způsobí, že z isocitrátu vzniká jen omezené množství α-ketoglutarátu a enzym navíc získává novou schopnost katalyzovat redukci α-ketoglutarátu na 2-hydroxyglutarát (schéma 1). Tato látka má toxický účinek na DNA. V současné době se předpokládá, že 2-hydroxyglutarát kompetitivně inhibuje aktivitu α-ketoglutarát dependentních dioxygenáz, mezi které patří i histon demetylázy [5]. Vlivem 2-hydroxyglutarátu tedy dochází k hypermetylaci histonů a tato hypermetylace pak neumožňuje transkripci důležitých tumor-supresorových genů, až dochází ke vzniku gliálního nádoru [6– 8].
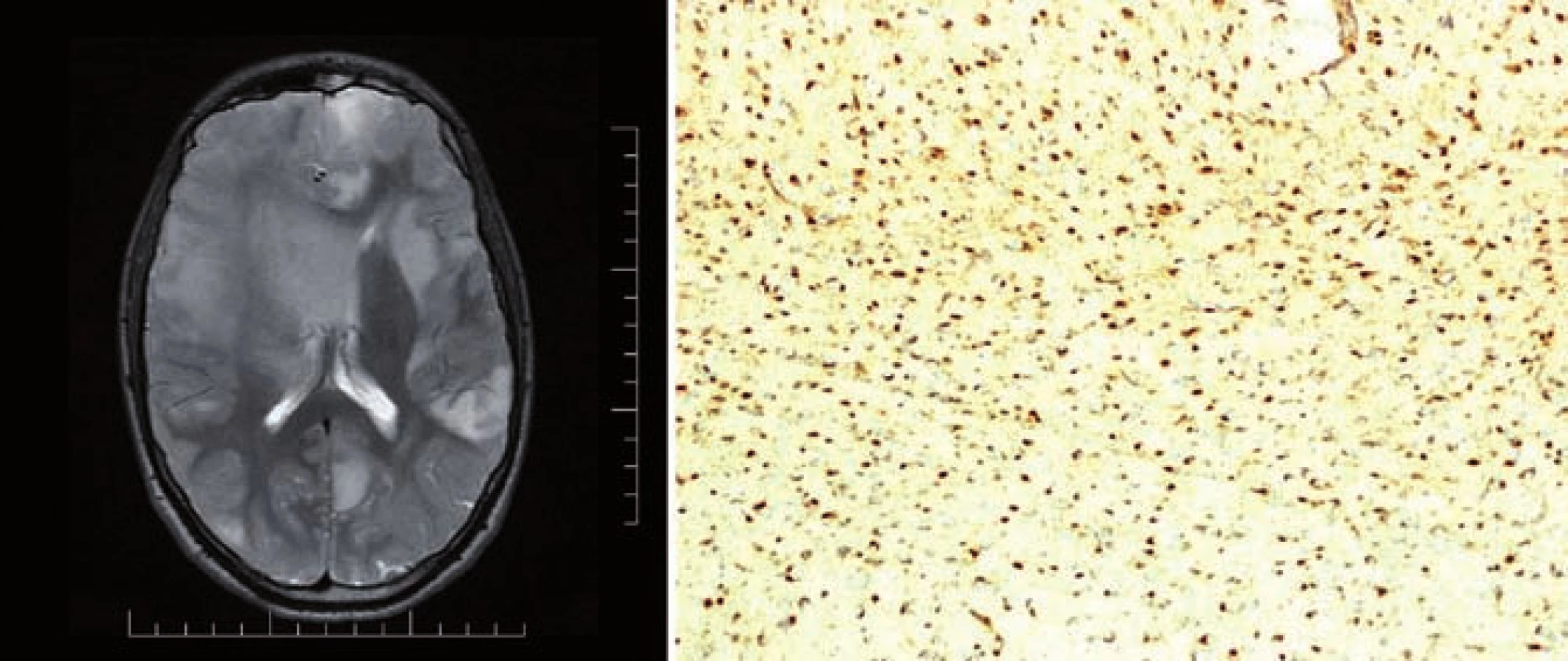
Mutace IDH1/ 2 byla následně objevena i u jiných malignit (akutní myeloidní leukemie, nádory prostaty, kartilaginozní nádory, akutní lymfoblastická leukemie, angioimunoblastický T lymfom). Roli IDH1/ 2 v tumorigenezi podporují dva důkazy. V prvním případě somatický mosaicizmus mutovaného IDH1 nebo IDH2 způsobuje kartilaginozní syndromy, Ollierovu chorobu a Maffucciho syndrom. Tyto syndromy jsou charakterizovány vznikem hemangiomů a chrupavkových nádorů, ale pacienti s tímto onemocněním mají i vyšší riziko vzniku gliálních nádorů (obr. 1). Druhým důkazem je zvýšená proliferace, tvorba kolonií a neschopnost diferenciace, pokud pomocí retroviru vložíme do normální buňky gen IDH1 nesoucí mutaci R132H [9,10]. V současné době bylo prokázáno, že tzv. G-CIMP fenotyp (glioma CpG island methylator phenotype) vzniká na podkladě mutace IDH, je vlastně výsledným produktem této mutace [11,12].
Velmi jednoduše můžeme gliální nádory rozdělit na tzv. IDH wild type (bez mutace) a na gliomy s mutací IDH. Mutace IDH je prognosticky příznivý faktor přežití především u dospělých pacientů s gliomy st. II–III. V dnešní době je diagnostika IDH mutací značně usnadněna možností imunohistochemické detekce IDH1 R132H (cca 95 % mutovaných IDH) [13]. V současnosti se hledá možnost, jak terapeuticky ovlivnit tyto gliomy: buď je cílem přímo mutaci nesoucí isocitrátdehydrogenáza (IDH1 R132H – AGI 5198, a IDH2 R140Q – AGI 6780), nebo snížení produkce 2-hydroxyglutarátu pomocí siRNA [9,14–16].
![Schéma 1. Vznik 2-hydroxyglutarátu a jeho vliv na DNA dioxygenázy (5-metyl-cytosin dioxygenázy, histon demetylázy). Převzato z [7].](https://pl-master.mdcdn.cz/media/image/884c7f6a59178dbe3450c5e7841dd902.jpg?version=1537795079)
1p/ 19q
Jedná se o chromozomální přestavbu, kterou typicky nacházíme u oligodendrogliomů. Jde o nebalancovanou translokaci krátkého ramene 1. chromozomu a dlouhého ramene 19. chromozomu. Tato přestavba byla nalezena již na konci 80. let minulého století a bylo potvrzeno několika studiemi, že její nositelé mají lepší prognózu onemocnění oproti těm, u kterých se nevyskytuje. Význam přítomnosti delece 1p a 19q tkví především ve faktu, že nádor, který ji nese, je chemosenzitivní. Pacienti, u nichž se mutace vyskytuje, tedy reagují daleko lépe na chemoterapii oproti těm, kteří tuto mutaci nemají. Tento rozdíl je vysoce statisticky významný a byl prokázán v několika randomizovaných studiích [17,18]. Při výskytu mutace IDH v kombinaci s kodelecí 1p/ 19q je prognóza nejpříznivější. Naopak pacienti, kteří mají wild type IDH, prakticky na chemoterapii nereagují a není rozdíl v přežití oproti skupině, která podstoupila pouze radioterapii [19]. Velmi často se současně s kodelecí 1p/ 19q vyskytuje i mutace genu CIC (19q13.2) a FUBP1 (1p31.1), u jiných typů gliomů se prakticky nevyskytují [1]. Jejich klinický význam nicméně zatím zůstává nejasný, jejich výskyt celkové přežití neovlivňuje [20].
TERT
Telomerázová reverzní transkriptáza (TERT) je funkční jednotkou enzymu telomerázy, která je odpovědná za zachování délky telomery při buněčném dělení (zamezuje zkracování telomer). Za fyziologických podmínek je telomeráza aktivní v dělících se zárodečných buňkách a u dospělého jedince v kmenových buňkách. U normálních buněk je její aktivita velmi nízká, ale u rychle se dělících tkání může být naopak velmi vysoká. Za patologických podmínek nacházíme zvýšenou telomerázovou aktivitu v nádorových buňkách [21]. V současné době je předpoklad, že existují dva způsoby, jak zachovat délku telomery v dělících se nádorových tkáních. Buď se jedná o kmenové nádorové buňky, kde je zachována telomerázová aktivita z podstaty toho, že se jedná o kmenovou buňku. Druhý mechanizmus, který je nezávislý na telomerázové aktivitě, je znám pod pojmem ALT (Alternative Lenghtening of Telomeres). ALT využívá kolem 10 % nádorů, v některých typech nádorů, např. gliálních nádorech, je však častější. Jeho příčinou jsou somatické mutace v promotoru TERT nebo mutace v genech DAXX nebo ATRX kódujících telomerázové vazebné proteiny [21].
Role mutace promotoru TERT byla popsána např. u melanomu. Nedávno ale byla objevena mutace TERT i u gliálních nádorů. Vyskytuje se především u oligodendrogliomů, ale i u GBM. Významně tuto hypotézu podporuje fakt, že mutace promotoru TERT se prakticky exkluzivně vylučuje s mutací ATRX. Buď je v nádorové tkáni přítomna jedna nebo druhá. Zde je možné vypozorovat, že u nádorů, které nesou kodeleci 1p/ 19q, se často vyskytuje mutace TERT promotoru [22]. Naopak u anaplastických gliomů, které nenesou kodeleci 1p/ 19q, je ve vysokém procentu nalezena mutace ATRX [23], viz dále.
ATRX
Role genu ATRX (alpha-thalassemia/mental retardation syndrome X-linked) v tumorigenezi gliomů byla prokázána teprve nedávno. V německé studii NOA-04 byla exprese tohoto genu zjišťována pomocí imunohistochemie u gliomů st. III. Ztráta exprese tohoto genu byla nalezena u 45 % anaplastických astrocytomů (AA), 27 % anaplastických oligoastrocytomů (AOA) a jen 10 % anaplastických oligodendrogliomů (AO). Byla nalezena prakticky pouze u nádorů s mutací IDH a zároveň se prakticky nikdy nevyskytovala s kodelecí 1p/ 19q [24]. Bylo prokázáno, že ztráta exprese ATRX u pacientů s anaplastickými nádory (v důsledku tzv. nonsense mutace tohoto genu) je prognosticky příznivý faktor přežití oproti těm pacientům, u nichž se tato ztráta exprese nevyskytuje [25]. Podstatnou roli v prognóze ale hraje u anaplastických oligoastrocytomů. V případě AOA a ztráty exprese ATRX odpovídá biologické chování nádoru anaplastickému astrocytomu. Pokud je naopak u AOA přítomna kodelece 1p/19q, pak se klinický průběh onemocnění podobá anaplastickému oligodendrogliomu; zde právě bývá často nalezena mutace promotoru TERT.
MGMT
Při metylaci DNA dochází k navázání metylové skupiny CH3 na 5’ pozici cytosinu. 5-metyl cytosin se většinou vyskytuje v rámci dinukleotidu CpG (název je odvozen od cytosin(C)-fosfát(p)-guanosin(G)). CpG dinukleotidy se v rámci savčího genomu vyskytují nerovnoměrně a odhaduje se, že 70–80 % jich je metylováno. V lidském genomu můžeme najít oblasti, které jsou na CpG dinukleotidy velmi bohaté. Právě tyto oblasti pak vytvářejí tzv. CpG ostrůvky (CpG islands), které se vyskytují v oblastech promotorů až poloviny lidských genů. DNA je pak metylována pomocí DNA metyltransferáz predilekčně v těchto promotorových oblastech, což vede k zastavení transkripce DNA. Tyto metylované CpG ostrůvky pak vytvářejí tzv. G-CIMP fenotyp.
U gliálních nádorů je často studovaným markerem metylace promotoru MGMT (O6- metylguanin-DNA metyltransferáza), která předurčuje, jak bude pacient reagovat na chemoterapii alkylačními činidly, např. temozolomidem. Temozolomid je schopný indukovat alkylaci DNA na pozici O6guaninu, tedy vytváří tzv. kovalentní adukty s DNA. Za fyziologických podmínek je toto poškození DNA opraveno enzymem MGMT a pacient na léčbu temzolomidem nereaguje. Metylace promotoru MGMT ale vede k zastavení transkripce MGMT, takže po chemoterapii alkylačními činidly nemůže být poškozená DNA opravena a pacient lépe odpovídá na léčbu.
Role metylace promotoru MGMT u GBM byla popsána již v roce 2005, kdy byla publikována práce, která prokázala vysokou účinnost chemoterapie temozolomidem u pacientů s GBM [26]. Pacienti, v jejichž nádorové tkáni byla tato metylace promotoru MGMT prokázána, reagovali lépe na podání temozolomidu oproti pacientům, u nichž tato metylace promotoru MGMT nebyla prokázána [27]. Metylace MGMT se vyskytuje cca ve 35 % GBM a 80 % LGG a je dalším prognosticky významným a nezávislým faktorem přežití pacientů s GBM.
EGFR
EGFRvIII je mutace EGFR, která se vyskytuje cca ve 30 % GBM. Nachází se často dohromady s overexpresí EGFR, kterou zjišťujeme u cca 50 % GBM [4]. EGFRvIII se typicky vyskytuje u primárních GBM, ale je vzácná u sekundárních GBM [28]. Mutovaný EGFRvIII je trvale aktivován díky ztrátě 267 aminokyselin z extracelulární domény a není schopen vázat žádný známý ligand. Díky jeho častému výskytu u primárních GBM a tomu, že se ve zdravé tkáni nenachází, je na něj soustředěna cílená imunoterapie. V současné době probíhá několik studií zaměřených právě na EGFRvIII (např. užití rindopepimutu) [29], které by mohly zlepšit přežití pacientů s GBM nesoucím tento typ mutace EGFR.
Jednonukleotidový polymorfizmus
V posledních letech bylo nalezeno několik vrozených znaků, jejichž nositelé mají vyšší riziko vzniku gliomů různých stupňů či se liší jejich prognóza. Jedná se o záměny bazí, tzv. jednonukleotidové polymorfizmy (Single Nucleotide Polymorphism; SNP), které se, na rozdíl od bodových mutací relativně často vyskytují i ve zdravé populaci. Ve většině těchto případů způsobí záměnu aminokyseliny v proteinovém řetězci, která má za následek drobnou modifikaci jeho funkce např. v důsledku jeho pozměněné sekundární struktury. V současné době známe více takovýchto polymorfních úseků, u kterých byl vysledován vztah k predispozici nebo prognóze pacientů s gliomy. Nejvýznamněji se v tuto chvíli jeví lokus 8q24.21 v blízkosti CCDC26, který byl analyzován u pacientů ve studii RTOG 9402. Jedná se o SNP rs55705857. Pacienti s AO, kteří nesou v tomto místě G alelu (GA, GG), mají podstatně lepší prognózu vůči pacientům bez G alely (homozygoti AA). Ve skupině pacientů s chemoterapií a radioterapií tento rozdíl činí 8,9 let oproti 3,3 let u pacientů – homozygotů AA. Zároveň pacienti s G alelou významně profitují z chemoterapie PCV (prokarbazin, CCNU, vinkristin), progression free survival (PFS) dosahuje 6,8 let ve srovnání s pacienty s G alelou, kteří podstoupili pouze radioterapii, zde PFS činí pouze 1,7 roku. Tato rozsáhlá molekulární analýza ukázala, že vrozená dispozice, histologický podtyp vč. statusu 1p/19q a celkové přežití jsou úzce provázány a že určité geneticky přenosné znaky mohou hrát v prognóze onemocnění podstatně významnější roli [19]. Bylo prokázáno, že právě SNP rs55705857 se významně častěji vyskytuje u oligodendrogliomů a astrocytomů nesoucích mutaci IDH1/ 2 [30]. Jiným takovým polymorfizmem je SNP rs34180180 v oblasti promotoru MGMT. Podobně jako u SNP rs55705857 i zde se ukazuje, že A alela je v tomto případě spojena s kratším celkovým přežitím, riziko smrti je u homozygotů GG o 82 % nižší než u homozygotů AA [31]. Promotor MGMT skrývá ještě jeden polymorfizmus významný pro metylaci MGMT a následnou terapeutickou odpověď na temozolomid. Jedná se o SNP rs16906252. Nositelé T alely v tomto místě exprimují podstatně méně MGMT, a tím lépe odpovídají na podání temozolomidu. Alela TC/TT se vyskytuje u 14–20 % pacientů. Medián přežití u jejích nositelů dosahuje 20 měsíců oproti 12,2 měsíců u pacientů s wild type alelou CC [32].
Byly provedeny ale i další genomové studie, které odhalily jiné polymorfní lokusy, které mohou ovlivňovat riziko vzniku a prognózu gliálních nádorů: 5p15.33 TERT, 7p11.2 EGFR, 8q24.21 CCDC26, 9p21.3 CDKN2A-CDKN2B, 11q23.3 PHLDB1 a 20q13.33 RTEL1 [33,34]. Ve studii porovnávající výskyt mutace IDH, kodeleci 1p/19q, mutaci promotoru TERT a 8 SNP (TERC (3q26), TERT (5p15), EGFR (7p12, 2 nezávislé oblasti), CCDC26 (8q24), CDKN2A nebo CDKN2B (CDKN2A/ B – 9p21), PHLDB1 (11q23), TP53 (17p13) a RTEL1 (20q13)) je ze všech těchto oblastí je i v této studii nejvýznamnější SNP rs55705857, který je spojen se zvýšeným rizikem vzniku gliomů s mutací IDH. PHLDB1 SNP rs498872 je spojen s gliomy, které nesou pouze IDH mutaci. Polymorfizmy TERC (rs1920116), TERT (rs2736100) a RTEL1 (rs6010620) naopak snižovaly riziko vzniku gliomu nesoucího pouze mutaci promotoru TERT. SNP CDKN2A/ B (rs4977756) se naopak vyskytuje častěji u tzv. triple-negativních gliomů (bez mutace IDH, TERT i kodelece 1p/ 19q) nebo u gliomů pouze s mutací TERT. SNP TP53 (rs78378222) je spojen se zvýšeným rizikem vzniku gliomů s IDH a TERT mutací. A nakonec SNP CCDC26 a TERT je spojen se zvýšeným rizikem vzniku proneurálních gliomů [22].
Koexistence a interakce různých molekulárně genetických znaků
V současné době se ukazuje, že prognóza pacientů s gliálními nádory do značné míry závisí na tom, jakou mutaci nebo chromozomální aberaci nese jejich nádorová tkáň nebo jakou vrozenou dispozici si nesou (SNP). Bylo opakovaně prokázáno, že nejlepší prognózu mají pacienti s mutací IDH1/ 2 a kodelecí 1p/ 19q, např. pacienti nesoucí tyto znaky ve studii RTOG 9402, kteří prodělali chemoterapii PCV, dosáhli mediánu celkového přežití 14,7 let. Pokud podstoupili pouze radioterapii, pak se v průměru dožili 6,8 let [19]. I pacienti, kteří mají mutaci IDH a zároveň mutaci promotoru TERT, mají velmi příznivou prognózu. Přitom samostatně se vyskytující mutace TERT je vyloženě nepříznivý prognostický faktor [22]. Pacienti s anaplastickým astrocytomem, kteří mají mutaci IDH1/ 2 a zároveň mutaci ATRX (k selhání léčby dochází až po 4,6 letech), mají lepší prognózu oproti těm, u kterých ke ztrátě exprese ATRX nedošlo (zde k selhání léčby dojde už po 2,7 letech) [25]. Podobně nepříznivou prognózu jako gliomy se samostatnou mutací TERT mají i tzv. wild type IDH gliomy st. II–III. Například pacienti s wild type IDH a bez kodelece 1p/ 19q dosahují průměrného přežití okolo 12 měsíců bez ohledu na podanou léčbu. V těchto případech se často vyskytují mutace a delece podobné jako u GBM a prognóza je podobná i dle našich zkušeností [35]. U GBM je nutné odlišit primární a sekundární GBM. Především sekundární GBM s mutací IDH má lepší prognózu než již výše zmíněný wild type IDH glioblastom. Nicméně důležitým faktorem i u GBM je právě zmíněná přítomnost mutace promotoru MGMT, která zvyšuje citlivost na alkylační cytostatikum temozolomid. Jak se ukazuje, i vrozená dispozice ve formě SNP může ovlivňovat odpověď na chemoterapii či tendenci ke vzniku jednoho nebo jiného nádoru.
GBM v naprosté většině vznikají klasickou cestou tumorigeneze, tedy aktivací onkogenů či inaktivací tumorsupresorů. Nejčastěji jsou v tkáni GBM porušeny tří nejznámější regulační cesty – cesty p53, cesty RB1 a cesty PI3K/ MAPK (schéma 2) [28]. Na základě rozsáhlé analýzy genomu několika set vzorků GBM bylo potvrzeno, že k porušení regulace p53 cesty dochází nejčastěji mutací genu TP53, v menší míře díky amplifikaci MDM2 a MDM4 genu. RB1 cesta bývá nejčastěji porušena delecí tumor-supresorového genu CDKN2A (méně i CDKN2B a CDKN2C), v menší míře se pak na dysregulaci této cesty podílí i amplifikace CDK4 a CDK6 či mutace RB1 genu. Cesta RTK/ PI3K/ MAPK je postižena především amplifikací EGFR, dále PDGFRA, méně i FGFR. K deregulaci může dojít také díky mutaci či deleci PTEN nebo PI3K (nejčastěji amplifikace PIK3C2B a mutace PIK3CA), event. mutací či delecí NF1. Poruchu regulace těchto tří cest nacházíme často dokonce současně v jednom nádoru (RTK/ PI3K/ MAPK v 88 %, p53 v 87 %, RB1 v 78 % nádorů). Určité genetické alterace se ale nevyskytují náhodně, nýbrž naopak v jakýchsi clusterech, takže je pak možné rozdělit GBM do čtyř typů. Nejčastější je klasický typ, ve kterém se často vyskytuje amplifikace EGFR, event. nejčastější subvarianta EGFRvIII společně se ztrátou chromozomu 10. Naopak zde často chybí mutace TP53. Rovněž bývá často přítomna delece 9p21 postihující CDKN2A a vedoucí k přerušení RB1 cesty. Dalším častým typem je proneurální typ GBM, který je typickým představitelem sekundárních GBM. Je přítomna mutace IDH1/ 2 a amplifikace PDGFRA, G-CIMP genotyp [12]. Rovněž se často vyskytuje mutace TP53. Třetím typem je mezenchymální typ GBM, kde bývá často přítomna delece NF1 a zvýšená exprese MET. Posledním typem je neurální typ GBM, kde je naopak genetických změn přítomno málo, je zvýšená exprese NEFL, GABRA1, SYT1 a SLC12A5 [4,36,37].
![Schéma 2. Postižení signálních cest p53, RB1 a PI3K/MAPK u glioblastoma multiforme. Převzato z [28].](https://pl-master.mdcdn.cz/media/image/4c76bd4565706202b36292c15b3d02a1.jpg?version=1537793845)
Závěrem lze říci, že při současných poznatcích o molekulárně genetických změnách gliálních nádorů je na místě u gliomů nižších stupňů provádět vyšetření statusu IDH1/ 2 a kodelece 1p/ 19q. U anaplastických astrocytomů je vhodné doplnit expresi ATRX, u oligodendrogliomů expresi TERT. V případě high grade gliomů (především astrocytární řady) je vhodné doplnit metylaci promotoru MGMT. Všechny tyto znaky sice zatím zásadně neovlivňují rozhodnutí o léčbě, ale slouží ke stanovení prognózy pacientů a z toho důvodu by měly být standardně vyšetřovány [38]. Do budoucna pak před námi stojí i výzvy ve smyslu vyšetřování bodových polymorfizmů v rámci preventivních vyšetření či před plánováním vlastní onkologické léčby.
Podpořeno z programového projektu Ministerstva zdravotnictví ČR s reg. č. NT 14253-3, projektu PRVOUK P27 a projektu MO 2012.
Veškerá práva podle předpisů na ochranu duševního vlastnictví jsou vyhrazena.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr. Filip Kramář, Ph.D.
Neurochirurgická a neuroonkologická klinika
1. LF UK a ÚVN – VFN Praha
U Vojenské nemocnice 1200
169 02 Praha 6
e-mail: filip.kramar@gmail.com
Přijato k recenzi: 8. 1. 2016
Přijato do tisku: 8. 2. 2016
Sources
1. Dubbink HJ, Atmodimedjo PN, Kros JM, et al. Molecular classification of anaplastic oligodendroglioma using next-generation sequencing: a report of the prospective randomized EORTC Brain Tumor Group 26951 phase III trial. Neuro Oncol 2015;18(3):388– 400. doi: 10.1093/ neuonc/ nov182.
2. Siegal T. Clinical impact of molecular biomarkers in gliomas. J Clin Neurosci Off J Neurosurg Soc Australas 2015;22(3):437– 44. doi: 10.1016/ j.jocn.2014.10.004.
3. Ostrom QT, Gittleman H, Fulop J, et al. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008– 2012. Neuro Oncol 2015;17(Suppl 4):iv1– 62. doi: 10.1093/ neuonc/ nov189.
4. Verhaak RG, Hoadley KA, Purdom E, et al. An integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR and NF1. Cancer Cell 2010;17(1):98. doi: 10.1016/ j.ccr.2009.12.020.
5. Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009;462(7274):739. doi: 10.1038/ nature08617.
6. Cohen AL, Holmen SL, Colman H. IDH1 and IDH2 mutations in gliomas. Curr Neurol Neurosci Rep 2013;13(5):345. doi: 10.1007/ s11910-013-0345-4.
7. Schiff D, Purow B. Neuro-oncology: five new things. Neurol Clin Pract 2013;3(4):326– 33. doi: 10.1212/ CPJ.0b013e3182a1ba35.
8. Megova M, Drabek J, Koudelakova V, et al. Isocitrate dehydrogenase 1 and 2 mutations in gliomas. J Neurosci Res 2014;92(12):1611– 20. doi: 10.1002/ jnr.23456.
9. Seltzer MJ, Bennett BD, Joshi AD, et al. Inhibition of glutaminase preferentially slows growth of glioma cellswith mutant IDH1. Cancer Res 2010;70(22):8981– 7. doi: 10.1158/ 0008-5472.CAN-10-1666.
10. Lu C, Ward PS, Kapoor GS, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012;483(7390):474– 8. doi: 10.1038/ nature10860.
11. Turcan S, Rohle D, Goenka A, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012;483(7390):479– 83. doi: 10.1038/ nature10866.
12. Noushmehr H, Weisenberger DJ, Diefes K, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010;17(5):510– 22. doi: 10.1016/ j.ccr.2010.03.017.
13. Camelo-Piragua S, Jansen M, Ganguly A, et al. Mutant IDH1-specific immunohistochemistry distinguishes diffuse astrocytoma from astrocytosis. Acta Neuropathol (Berl) 2010;119(4):509– 11. doi: 10.1007/ s00401-009-0632-y.
14. Li L, Paz AC, Wilky BA, et al. Treatment with a Small Molecule Mutant IDH1 Inhibitor Suppresses Tumorigenic Activity and Decreases Production of the Oncometabolite 2-Hydroxyglutarate in Human Chondrosarcoma Cells. PloS One 2015;10(9):e0133813. doi: 10.1371/ journal.pone.0133813.
15. Suijker J, Oosting J, Koornneef A, et al. Inhibition of mutant IDH1 decreases D-2-HG levels without affecting tumorigenic properties of chondrosarcoma cell lines. Oncotarget 2015;6(14):12505– 19.
16. Kernytsky A, Wang F, Hansen E, et al. IDH2 mutation-induced histone and DNA hypermethylation is progressively reversed by small-molecule inhibition. Blood 2015;125(2):296– 303. doi: 10.1182/ blood-2013-10-533604.
17. Cairncross JG, Ueki K, Zlatescu MC, et al. Specific Genetic Predictors of Chemotherapeutic Response and Survival in Patients With Anaplastic Oligodendrogliomas. J Natl Cancer Inst 1998;90(19):1473– 9.
18. Cairncross G, Wang M, Shaw E, et al. Phase III Trial of Chemoradiotherapy for Anaplastic Oligodendroglioma: Long-Term Results of RTOG 9402. J Clin Oncol 2013;31(3):337– 43. doi: 10.1200/ JCO.2012.43.2674.
19. Cairncross JG, Wang M, Jenkins RB, et al. Benefit From Procarbazine, Lomustine, and Vincristinein Oligodendroglial Tumors Is Associated With Mutationof IDH. J Clin Oncol 2014;32(8):783– 90. doi: 10.1200/ JCO. 2013.49.3726.
20. Cancer Genome Atlas Research Network, Brat DJ, Verhaak RG, et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N Engl J Med 2015;372(26):2481– 98. doi: 10.1056/ NEJMoa1402121.
21. Killela PJ, Reitman ZJ, Jiao Y, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci U S A 2013;110(15):6021– 6. doi: 10.1073/ pnas.1303607110.
22. Eckel-Passow JE, Lachance DH, Molinaro AM, et al. Glioma Groups Based on 1p/ 19q, IDH, and TERT Promoter Mutations in Tumors. N Engl J Med 2015;372(26): 2499– 508. doi: 10.1056/ NEJMoa1407279.
23. Reuss DE, Sahm F, Schrimpf D, et al. ATRX and IDH1-R132H immunohistochemistry with subsequent copy number analysis and IDH sequencing as a basis for an “integrated” diagnostic approach for adult astrocytoma, oligodendroglioma and glioblastoma. Acta Neuropathol (Berl) 2015;129(1):133– 46. doi: 10.1007/ s00401-014-1370-3.
24. Kannan K, Inagaki A, Silber J, et al. Whole-exome sequencing identifies ATRX mutation as a key molecular determinant in lower-grade glioma. Oncotarget 2012;3(10):1194– 203.
25. Wiestler B, Capper D, Holland-Letz T, et al. ATRX loss refines the classification of anaplastic gliomas and identifies a subgroup of IDH mutant astrocytic tumors with better prognosis. Acta Neuropathol (Berl) 2013;126(3):443– 51. doi: 10.1007/ s00401-013-1156-z.
26. Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N Engl J Med 2005;352(10):987– 96. doi: 10.1056/ NEJMoa043330.
27. Hegi ME, Diserens AC, Gorlia T, et al. MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma. N Engl J Med 2005;352(10):997– 1003. doi: 10.1056/ NEJMoa043331.
28. Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008;455(7216):1061– 8. doi: 10.1038/ nature07385.
29. Zussman BM, Engh JA. Outcomes of the ACT III Study: Rindopepimut (CDX-110) Therapy for Glioblastoma. Neurosurgery 2015;76(6):N17. doi: 10.1227/ 01.neu.0000465855.63458.0c.
30. Jenkins RB, Xiao Y, Sicotte H, et al. A low-frequency variant at 8q24.21 is strongly associated with risk of oligodendroglial tumors and astrocytomas with IDH1 or IDH2 mutation. Nat Genet 2012;44(10):1122– 5. doi: 10.1038/ ng.2388.
31. Fogli A, Chautard E, Vaurs-Barrière C, et al. The tumoral A genotype of the MGMT rs34180180 single-nucleotide polymorphism in aggressive gliomas is associated with shorter patients’ survival. Carcinogenesis 2015;37(2):169– 76. doi: 10.1093/ carcin/ bgv251.
32. Rapkins RW, Wang F, Nguyen HN, et al. The MGMT promoter SNP rs16906252 is a risk factor for MGMT methylation in glioblastoma and is predictive of response to temozolomide. Neuro Oncol 2015;17(12):1589– 98. doi: 10.1093/ neuonc/ nov064.
33. Shete S, Hosking FJ, Robertson LB, et al. Genome-wide association study identifies five susceptibility loci for glioma. Nat Genet 2009;41(8):899– 904. doi: 10.1038/ ng.407.
34. Sanson M, Hosking FJ, Shete S, et al. Chromosome 7p11.2 (EGFR) variation influences glioma risk. HumMol Genet 2011;20(14):2897– 904. doi: 10.1093/ hmg/ ddr192.
35. Kramar F, Zemanova Z, Michalova K, et al. Cytogenetic analyses in 81 patients with brain gliomas: correlation with clinical outcome and morphological data. J Neurooncol 2007;84(2):201– 11. doi: 10.1007/ s11060-007-9358-7.
36. Brennan CW, Verhaak RGW, McKenna A, et al. The somatic genomic landscape of glioblastoma. Cell 2013;155(2):462– 77. doi: 10.1016/ j.cell.2013.09.034.
37. Mao H, LeBrun DG, Yang J, et al. Deregulated Signaling Pathways in Glioblastoma Multiforme: Molecular Mechanisms and Therapeutic Targets. Cancer Invest 2012;30(1):48– 56. doi: 10.3109/ 07357907.2011.630050.
38. Kalita O, Kramář F, Neumann E, et al. Současný stav léčby anaplastických gliomů v České republice. Cesk Slov Neurol N 2015;78/ 111(3):306– 16.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2016 Issue 4
Most read in this issue
- Neurorehabilitace senzomotorických funkcí po poranění míchy
- Tři časy Testu kreslení hodin hodnocené BaJa skórováním u časné Alzheimerovy nemoci
- Genetické a epigenetické faktory podmiňující vznik a prognózu mozkových gliomů – souhrn současných poznatků
- Měření vrstvy nervových vláken sítnice u pacientů s Alzheimerovou chorobou