
Překrývání neurodegenerativních demencí
Overlapping of Neurodegenerative Dementias
Neuropathological diagnostic criteria of neurodegenerative disorders are based on the presence of specific lesions in the brain tissue that correlate with clinical symptoms. Concomitant neurodegenerative disorders correspond to a combination of two (or more) different fully developed diseases in one patient. Concomitant neurodegenerative pathology stays for the presence of a definite neurodegeneration and deposits specific for another, but not fully developed, disease. Frequent overlaps include Alzheimer’s disease (AD) and alpha-synuclein inclusions. In AD, protein TDP-43 may co-aggregate but it is not clear whether this is an atypical but isolated AD, or an overlap of AD with early frontotemporal lobar degeneration. Comorbidities of AD and tauopathies are relatively rare. In Creutzfeldt-Jakob disease, concomitant AD or Lewy body dementia may occur. A combination of vascular pathology with a primary neurodegeneration (mostly Alzheimer’s disease or Lewy body dementia) is historically called mixed dementia. Overlap of neuropathologically confirmed neurodegenerations may lead to atypical and unusual clinical presentations, illustrated with examples and references to published case reports from our patient cohort.
Key words:
neurodegeneration – Alzheimer’s disease – tauopathy – TDP-43 protein – synucleinopathy – mixed dementia
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Authors:
Z. Rohan 1; R. Matěj 1–3
![]() ; R. Rusina 3,4
; R. Rusina 3,4
Authors‘ workplace:
Oddělení patologie a molekulární medicíny, Thomayerova nemocnice, Praha
1; Ústav patologie, 3. LF UK v Praze
2; Neurologická klinika a Centrum klinických neurověd, 1. LF UK a VFN v Praze
3; Neurologické oddělení, Thomayerova nemocnice, Praha
4
Published in:
Cesk Slov Neurol N 2015; 78/111(6): 641-648
Category:
Review Article
doi:
https://doi.org/10.14735/amcsnn2015641
Overview
Neuropatologická diagnostická kritéria neurodegenerací jsou založena na průkazu specifických změn v parenchymu korelujících s klinickým obrazem. Souběžná neuropatologicky definovaná onemocnění odpovídají kombinaci dvou (i více) odlišných, plně rozvinutých postižení u téhož pacienta. U souběžné patologie více entit v ohraničených oblastech mozku vedle primární nozologické jednotky nacházíme zároveň depozita proteinu specifického pro jinou neurodegeneraci. Častá je kombinace Alzheimerovy nemoci (AN) s inkluzemi alfa‑synukleinu. U AN se může zároveň ukládat i protein TDP ‑ 43, není ale jasné, zda se jedná o atypicky probíhající AN nebo kombinaci atypické AN s frontotemporální lobární degenerací. Komorbidity AN a tauopatií jsou relativně vzácné. U pacientů s Creutzfeldtovou ‑ Jakobovou nemocí lze diagnostikovat doprovodnou AN nebo demenci s Lewyho tělísky. Kombinace vaskulární patologie s primární neurodegenerací (nejčastěji AN nebo demencí s Lewyho tělísky) se historicky označuje jako smíšená demence. Kombinace neuropatologicky potvrzených neurodegenerací může vést k atypickým klinickým projevům, které ilustrujeme na příkladech s odkazy na publikované kazuistiky ze souboru našich pacientů.
Klíčová slova:
neurodegenerace – Alzheimerova nemoc - tauopatie – protein TDP-43 – synukleinopatie – smíšená demence
Úvod
Neurodegenerace jsou důsledkem postupného zániku specifických skupin neuronů. Patofyziologickou podstatou je ukládání určitého specifického, pro dané onemocnění charakteristického proteinu (např. beta-amyloidu, tau proteinu nebo alfa‑synukleinu) v mozkové tkáni v kombinaci s obecnými mechanizmy apoptózy a autofagie (řízené smrti buňky). V současné době tedy chápeme neurodegenerativní onemocnění jako specifické proteinopatie, kdy platí, že definitivní diagnóza je vždy neuropatologická.
Klinický obraz daného neurodegenerativního onemocnění je výsledkem selektivního postižení určité neuronální subpopulace a u řady onemocnění časoprostorovou progresí patologie daného proteinu (Alzheimerova nemoc (AN), demence s Lewyho tělísky (DLB), některé tauopatie). Správné rozpoznání neurodegenerací během života pacientů je i v současné době velmi obtížné. Klinicky se diagnóza určuje na úrovni „možné (possible)“ a „pravděpodobné (probable)“, zatímco „potvrzená (definite)“ diagnóza je pouze neuropatologická – na podkladě imunohistochemického průkazu typického nativního nebo patologicky modifikovaného proteinu (markeru) ve specifických oblastech mozku.
Obecným rysem neurodegenerací je korelace mezi postupně se horšícími klinickými projevy onemocnění a časoprostorovou progresí neuropatologických změn (depozita specifických proteinů a numerická atrofie – zánik neuronů) v mozkové tkáni.
Hlavní klinické a neuropatologické aspekty jednotlivých neurodegenerací jsou shrnuty v tab.1 a obr. 1.
![Srovnání typických klinických projevů a neuropatologických markerů neurodegenerativních demencí [34].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/2097527768c9ec20ed30969e357878c7.jpg)
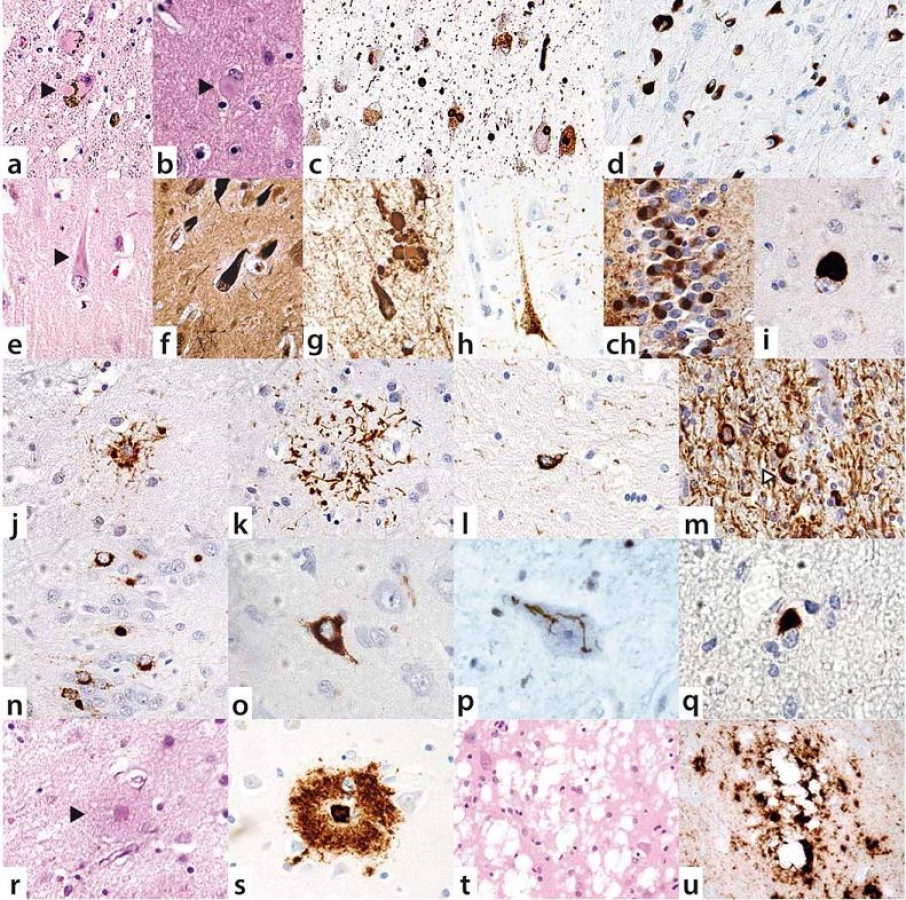
V poslední době se ukazuje, že neurodegenerativní onemocnění se nemusí vyskytovat izolovaně, ale u téhož pacienta se mohou překrývat různé etiopatogenetické jednotky. Komorbidity vedou k větší tíži postižení mozkového parenchymu, a mohou tedy mít rychlejší průběh a atypické klinické prezentace, rovněž horší odpovídavost na léčbu a méně příznivou prognózu.
Skutečnost, že překrývání různých neurodegenerací nemusí být excesivně vzácné, nýbrž že se lze s ním setkat i v běžné praxi, ilustrujeme na konkrétních příkladech s odkazy na klinicky dokumentované a neuropatologicky ověřené kazuistiky ze souboru našich pacientů, publikované v odborné literatuře.
Klinické a neuropatologické aspekty hlavních neurodegenerací
Alzheimerova nemoc
Markerem AN je přítomnost neuritických či difúzních plak imunoreaktivních s protilátkou proti beta-amyloidu a nález inkluzí patologicky konformovaného tau proteinu v hipokampu a korových oblastech.
Typická forma AN se v důsledku časného postižení transtentoriální oblasti, parahipokampálního závitu a hipokampu projevuje zprvu jako progredující amnestický syndrom s alterací epizodické paměti. Později se patologické změny šíří dále temporálně i extratemporálně, zprvu parietálně, později i frontálně. Tomu odpovídá rozvoj dalších klinických příznaků – poruch řeči, zrakově‑konstruktivních dysfunkcí, exekutivního postižení a další.
Neuropatologická, tzv. ABC klasifikace, navržená National Institute on Aging ‑ Alzheimer’s Association (NIA ‑ AA) hodnotí distribuci i denzitu amyloidových plak (A – Amyloid, C – CERAD) a tau proteinu (B – Braak). Výsledkem je pravděpodobnost (nízká ‑ střední ‑ těžká) podílu neuropatologických změn na pozorovaném kognitivním deficitu [1].
Kromě výše popsané typické formy AN se v přibližně v 20 – 25 % případů může AN projevit ve svých atypických formách [2,3]. Jednou je tzv. limbic ‑ predominant forma s relativně pokročilou atrofií mediálních temporálních struktur, která není doprovázena atrofií kortikálních asociačních oblastí odpovídající stupni kognitivního postižení. V klinickém obrazu převládá časné postižení paměti hipokampálního typu, které zůstává dlouho izolováno a později se přidává porucha exekutivních funkcí, zatímco řeč a zrakově‑prostorové funkce zůstávají dlouho ušetřeny.
Druhou atypickou formou je tzv. hipokampus ‑ šetřící (hippocampal ‑ sparing) forma AN, u které je na rozdíl od limbic ‑ predominant formy korová atrofie relativně pokročilejší oproti atrofii mediálních temporálních struktur. U těchto pacientů je postižení epizodické paměti jen mírné, ale nepaměťové kognitivní funkce (řeč, exekutivní a zrakově‑prostorové funkce) jsou výrazně alterovány již na počátku onemocnění.
Mediální temporální atrofie (MTA) tak v případě hipokampus ‑ šetřící formy AN může, zvláště u starších osob, spadat i do pásma mírné či věku přiměřené MTA, a značně tak může znesnadnit diagnostiku AN, a tím významně ovlivnit prospektivní diagnostiku nejen v rámci klinické praxe, ale i lékových či biomarkerových studií.
Synukleinopatie
Mezi synukleinopatie se řadí Parkinsonova nemoc, DLB, multisystémová atrofie (MSA) a další vzácnější jednotky. Hlavním příznakem je parkinsonský syndrom charakterizovaný hypokinézou, rigiditou, třesem a posturální instabilitou. Další přidružené projevy mohou zahrnovat kognitivní postižení, mozečkovou nebo autonomní dysfunkci [4].
Pro synukleinopatie jsou typické intracelulární inkluze patologicky konformovaného proteinu alfa‑synukleinu. Základní typy těchto inkluzí jsou Lewyho tělíska, Lewyho neurity a oligodendrogliální inkluze (tzv. Pappovy ‑ Lantosovy inkluze).
Lewyho tělíska se mohou vyskytovat v klasické kmenové formě (eozinofilní, s periferním projasněním) a v méně výrazné korové formě (eozinofilní, bez halo). Lewyho neurity jsou výběžky neuronů s patologicky agregovaným alfa‑synukleinem viditelné až po použití specifických protilátek (proti alfa‑synukleinu, ubikvitinu atd). Pappovy ‑ Lantosovy inkluze jsou „plaménkové“ inkluze v oligodendroglii viditelné v impregnaci solemi stříbra a v imunohistochemickém průkazu s protilátkou proti alfa‑synukleinu.
Tauopatie
Tato onemocnění jsou zapříčiněna abnormálním metabolizmem tau proteinu (z anglického „tubulin associated unit“) a jeho intraneuronálním ukládáním či depozity v gliových elementech.
Tauopatie jsou významnou součástí frontotemporálních lobárních degenerací (FTLD), skupiny onemocnění velmi heterogenní z pohledu klinického i etiopatogenetického.
Nejčastější jsou tau pozitivní frontotemporální demence (vč. Pickovy nemoci) s postižením frontálních funkcí s časnými poruchami chování, osobnostními změnami a poruchou paměti v důsledku hipokampálních lézí.
Nonfluentní/ agramatická varianta primární progresivní afázie se projevuje zpočátku izolovanou výrazně narušenou produkcí řeči, která postupně progreduje do obrazu těžké frontální demence.
U progresivní supranukleární obrny (PSP) převažuje atypický parkinsonský syndrom s dominancí posturální instability a četnými pády již v počátku nemoci, objevuje se supranukleární obrna pohledu a poruchy behaviorální a kognitivní.
Pro kortikobazální degeneraci je příznačná demence s časnou apraxií, afázií a frontální symptomatikou v kombinaci s výrazně asymetrickým parkinsonským syndromem nereagujícím na dopaterapii, „fenoménem cizí ruky“ a asymetrickými dystonickými projevy s myokloniemi.
Mezi tauopatie patří i vzácné, převážně neuropatologicky definované nemoci s neujasněným klinickým obrazem: nemoc s argyrofilními zrny (AGD), tauopatie bílé hmoty s globulárními gliálními inkluzemi a věkově vázaná primární tauopatie (Primary Age ‑ Related Tauopathy; PART), pod kterou byly recentně zahrnuty promiskue užívané pojmy senilní demence s tangles, tangle ‑ only varianta AN či demence s tangles popisující nález neurofibrilární patologie tau proteinu zejména v limbických oblastech při absenci beta-amyloidových plak s klinicky variabilním kognitivním deficitem [5,6].
Charakteristickým rysem tauopatií je predominance jedné ze dvou základních izoforem tau proteinu (4R či 3R). Pickova nemoc je typický zástupce 3R ‑ tauopatií, progresivní supranukleární obrna (PSP), kortikobazální degenerace nebo AGD jsou pak 4R ‑ tauopatie [7].
Proteinopatie TDP‑43
Inkluze pTDP ‑ 43 (hyperfosforylované formy proteinu TDP ‑ 43 – transactive response DNA binding protein 43) nacházíme v neuronech i glii v předních rozích míšních a motorickém, pre‑/ frontálním a temporálním kortexu, hipokampu, subkortikálních jádrech a kmenových strukturách i mozečku [8].
Depozita pTDP ‑ 43 jsou nejčastější příčinou tau negativních frontotemporálních demencí (klinický obraz je neodlišitelný od symptomatologie Pickovy nemoci), některých forem primárních progresivních afázií (sémantická varianta) a mají klíčovou roli u frontotemporální lobární degenerace s onemocněním motorického neuronu (FTLD ‑ MND) – asociace amyotrofické laterální sklerózy (ALS) s frontálním typem demence [9].
Na druhou stranu až ve 30 % případů lze nalézt TDP ‑ 43 pozitivní inkluze v amygdale a hipokampu kognitivně zdravých osob, a to i u osob nad 90 let věku [10]. Význam těchto depozit není jasný.
Prionová onemocnění
Typickým klinickým obrazem prionových onemocnění, způsobených ukládáním patologicky změněného prionového proteinu (PrPSc) do mozkové tkáně s postupujícím zánikem neuronů, je u nejčastější formy – Creutzfeldtovy ‑ Jakobovy nemoci rychle progredující demence s dalšími klinickými abnormalitami (myoklonus, zrakové nebo mozečkové projevy, pyramidové/ extrapyramidové postižení a akinetický mutizmus).
Klasickou neurohistologickou trojicí změn při prionových nemocech je spongiformní dystrofie, numerická atrofie neuronů a reaktivní astroglióza. Imunohistochemické vyšetření užívá různé typy protilátek ozřejmujících výskyt patologicky konformovaných prionů ve tkáni [11].
Překrývání neurodegenerací
Neuropatologická verifikace má u neurodegenerativních onemocnění zásadní význam. Až ve 20–40 % případů může být totiž stanovená klinická diagnóza nesprávná, tedy nezřídka je diagnostikována např. Alzheimerova nebo Parkinsonova nemoc, přestože pacient trpí úplně jiným onemocněním [12].
V poslední době se navíc ukazuje, že společný výskyt více markerů definujících různé neurodegenerace není vzácný; je naopak poměrně častý, to zejména úměrně se stoupajícím věkem. Můžeme se tak setkat s typickým nálezem určitého onemocnění provázeným ohraničenými depozity jiného onemocnění, které však není plně rozvinuté – nebo i s opravdovou komorbiditou, současným výskytem dvou odlišných nozologických jednotek [13,14].
Lze tak rozlišovat tři možné stavy (tab. 2):

- Souběžná neurodegenerativní onemocnění – komorbidity: dvě i více plně vyvinutá neurodegenerativní onemocnění splňují daná diagnostická kritéria (např. AN v kombinaci s multisystémovou atrofií či DLB) [15].
- Souběžná neurodegenerativní patologie: v terénu primární, neuropatologicky i klinicky definované neurodegenerace (např. Pickovy nemoci) lze nalézt neuropatologické změny charakteru inkluzí či depozit jiného proteinu (např. alfa‑synukleinu v amygdale) v ohraničeném rozsahu, nesplňující však daná diagnostická kritéria pro další onemocnění per se.
- Smíšená demence (smíšená neuropatologie): pojem užívaný pro současný nález vyvinutého neurodegenerativního onemocnění a cerebrovaskulární patologie. Typicky se jedná o vaskulární demenci (dříve Binswangerova nemoc) spojenou s AN, v širším slova smyslu však může jít o kombinaci s jakoukoliv primární neurodegeneraci. Tíže a frekvence cerebrovaskulární patologie rovněž výrazně narůstá s věkem.
Alzheimerova nemoc a synukleinopatie
Současný výskyt markerů AN (neuritické plaky a neurofibrilární klubka, tangles) a patologie alfa‑synukleinu (Lewyho tělíska a Lewyho neurity) může mít dvojí charakter.
Pokud vedle plně rozvinutých alzheimerovských změn jsou přítomna i depozita alfa‑synukleinu v izolovaných oblastech, nejčastěji v limbických strukturách, typicky v amygdale a v gyrus cinguli (ale chybí v kmenových jádrech), jedná se většinou o AN se souběžnou alfa‑synukleinopatií, přičemž hlavní podíl na klinickém obraze lze připsat AN spolu s (prakticky vždycky identifikovatelnými) vaskulárními změnami.
Komorbidita AN a synukleinopatie se vyznačuje nálezem rozvinuté alzheimerovské patologie (potvrzená „definite“ AN v ABC klasifikaci NIA ‑ AA) a rozsáhlých kortikálních i subkortikálních depozit alfa‑synukleinu (potvrzená „definite“ DLB podle Braaka event. McKeitha – nebo potvrzená „definite“ multisystémová atrofie). Závěry recentních studií navíc poukazují na možnost vzájemného vztahu mezi patologií tau proteinu, beta amyloidu a alfa‑synukleinu, který může vést k potenciaci neuropatologických a zřejmě i klinických změn [16,17].
V těchto případech klinická manifestace může mít atypické charakteristiky, odlišné od typického průběhu DLB a AN, nebo MSA a AN, např. rychle progredující demence s parkinsonizmem a závažnými poruchami chování připomínající frontotemporální demenci v prvním případě [18] nebo rozvoj těžké demence v rámci nezvykle rychle postupující MSA [15].
Alzheimerova nemoc a tauopatie
Komplexita patologie tau proteinu je zřejmá u kombinací AN s tauopatiemi. Lze vysledovat určitou podobnost mezi tauopatiemi a AN – např. u klasické formy PSP podobně jako u AN koreluje klinická manifestace s progresí ukládání depozit tau proteinu v mozku.
Tauopatie jsou klinicky a morfologicky velmi heterogenní – neuropatologicky se jedná o přítomnost 3R či 4R inkluzí tau v neuronech a glii a jejich morfologii a distribuci v rámci kmenových, mozečkových či subkortikálních a kortikálních struktur.
Při splnění přesných kritérií lze pak uvést diagnózu specifické tauopatie (FTD, PSP, kortikobazální degenerace), nicméně velká část případů má morfologicky ne zcela vyvinutý charakteristický obraz či jsou variabilně postiženy další, pro danou tauopatii atypické, oblasti mozku. Tyto případy spadají do široké skupiny tzv. komplexních tauopatií, jednotek definovaných neuropatologicky a s velmi rozmanitými klinickými projevy (kognitivní alterace, extrapyramidová či mozečková symptomatika atd.).
Komorbidita AN a Pickovy nemoci se může projevovat jako amnestická demence s progresivní afázií a apraxií, progredující do těžké frontální demence [19]. Komorbidita progresivní supranukleární obrny a AN může nápadně připomínat synukleinopatie, např. DLB i multisystémovou atrofii [20].
Souběžná neurodegenerativní patologie kombinující AN s argyrofilní demencí je poměrně častá, její incidence roste s věkem (stejně jako incidence izolované AGD). Podíl AGD na kognitivní alteraci je sporný (tím spíše, že AGD je zatím pouze neuropatologicky definované onemocnění, u něhož momentálně není znám typický klinický průběh). U hraničních stadií AN (limbické stadium; NIA ‑ AA ABC intermediate či low) může přítomnost AGD být rozhodující faktor pro klinickou manifestaci mírné kognitivní poruchy nebo syndromu demence [21–23].
Alzheimerova nemoc a proteinopatie TDP‑43
Souběžná neurodegenerativní patologie TDP ‑ 43 inkluzí u prokázané „definite“ AN je relativně častá.
Až u 50 % případů AN lze pTDP ‑ 43 inkluze nalézt prakticky vždy v amygdale, v hipokampálních strukturách, zejména ve spojitosti s hipokampální sklerózou (zde se však nejedná o hipokampální sklerózu ischemické etiologie asociovanou s epilepsií, ale o hipokampální sklerózu způsobenou degenerací pyramidových neuronů cornu ammonis v rámci primární neurodegenerace, např. AN), v subkortikálních jádrech, ve kmeni i v korových oblastech. Časně se TDP ‑ 43 depozita objevují v amygdale a s další progresí AN postupují přes hipokampální struktury dále do podkorových a korových oblastí [24].
Recentní studie ukázala, že AN se současným nálezem TDP ‑ 43 inkluzí bez symptomatologie odpovídající FTD či MND/ ALS má horší kognitivní profil a jiný charakter i míru korové atrofie než u alzheimerovských pacientů bez TDP ‑ 43 inkluzí. Klinický obraz navíc pozitivně koreloval s mírou patologie TDP ‑ 43 [25]. Patologie TDP ‑ 43 tedy může spolu s patologickými změnami v rámci AN synergisticky potencovat kognitivní deterioraci, podobně jako je tomu u vaskulární patologie či AGD.
Na druhou stranu, přítomnost TDP ‑ 43 v bazálních gangliích a substantia nigra nevedl k významnějšímu výskytu parkinsonských příznaků či FTD [26].
Komorbidita AN a ALS s demencí (FTLD ‑ MND) je v publikované literatuře považována za vzácnější, klinicky se může projevit poměrně rychle postupujícím onemocněním motorického neuronu s demencí převážně amnestického rázu a temporální atrofií na MR [27]. Při pečlivém neuropatologickém vyšetřování pacientů s ALS s kognitivním deficitem však byla přítomnost alzheimerovských změn různé tíže ve skupině námi analyzovaných případů relativně častá (nepublikované pozorování).
Prionový protein a komorbidity
Přítomnost prionového onemocnění nevylučuje možnost další souběžné neurodegenerace, která mohla mít vliv na stav pacienta před vlastním nástupem projevů prionového onemocnění. Relativně častější je u genetických forem CJN, zejména u specifické formy s mutací E200K [28].
Nejčastější souběžnou neuropatologií je patologie tau proteinu, která může, vč. přítomnosti difuzních či neuritických plak, splnit diagnostická kritéria pro diagnózu PART (Primary Age ‑ Related Tauopathy – věkově vázaná primární tauopatie) nebo AN, většinou však jen v jejím entorinálním či limbickém stadiu (pásmo NIA ‑ AA low a intermediate).
Jako u dalších neurodegenerativních onemocnění nelze ani u prionových onemocnění prakticky vyloučit možnost kombinace dvou neurodegenerací či souběžné neuropatologie. Popisovány jsou kombinace Creutzfeldtovy ‑ Jakobovy nemoci s AN, DLB nebo dokonce MSA [29 – 31].
Smíšená demence
Pravděpodobně nejčastější kombinací neurodegenerativních onemocnění sensu lato je kombinace vaskulární patologie a jiného, primárního, neurodegenerativního onemocnění, zejména AN. Pravděpodobnost výskytu cerebrovaskulárních změn navíc narůstá s věkem.
Pojem vaskulární patologie v sobě z neuropatologického hlediska zahrnuje celou řadu lézí, od makroskopicky zcela jasně identifikovatelných lézí s odpovídajícím klinickým korelátem až po mikroskopické léze, jejichž význam je nejistý. Právě díky morfologické a klinické rozmanitosti těchto lézí jednotná neuropatologická klasifikace a staging prakticky neexistují, a neuropatolog se tak omezuje na pouhý popis nalezených změn [32,33]. Přehled základních typů vaskulární patologie je uveden v tab. 3.
V ideálním případě je jedna ze složek smíšené demence v plně vyvinutém stadiu a druhá je spíše doprovodná – např. AN v neokortikálním stadiu (v ABC pásmu high dle NIA ‑ AA) kombinovaná s nečetnými drobnými lakunami v oblasti bazálních ganglií či naopak rozsáhlá vaskulární patologie bazálních ganglií s nálezem Lewyho patologie v kmenových strukturách, která sama o sobě nemůže extrapyramidovou symptomatiku vyvolat.
Závažnost ložiskových vaskulárních změn lze částečně posoudit jejich lokalizací a orientačním stanovením jejich celkového objemu. Velmi hrubě orientačně lze říci, že izolované léze, jejichž kombinovaný objem přesahuje 20 ml, mohou být příčinou kognitivní alterace, přičemž léze nad 100 ml kombinovaného objemu pak mohou být odpovědné za plně vyvinutý syndrom demence.
Rychlá progrese demence může provázet případy AN kombinované s akutním vaskulárním inzultem (který může mít jakoukoliv podobu uvedenou v tab. 2), u nichž je pak častý likvorologický obraz extenzivní neuronální léze s velmi zvýšenými hodnotami celkového tau proteinu a pozitivitou proteinu 14 - 3 - 3, což může připomínat až obraz prionového onemocnění.
Pro diagnózu izolované vaskulární demence je však třeba definitivně vyloučit přítomnost dalších neurodegenerací. Je zásadní zhodnotit, do jaké míry se jednotlivé složky smíšené demence podílely na klinickém obraze. Přesto i při znalosti výsledků dalších vyšetření je často problematické k definitivní diagnóze dojít a někdy ani podrobná klinicko‑patologická korelace klinickou symptomatologii uspokojivě neobjasní.
Závěr
Kombinace dvou i více neurodegenerativních onemocnění jsou častější, než se dříve soudilo, i když zatím nejsou dostupná přesnější epidemiologická data. Překrývání neurodegenerací může být příčinou rychlejšího průběhu i někdy velmi atypických klinických obrazů u řady pacientů. Je proto třeba zdůraznit nutnost provádění detailního neuropatologického vyšetření mozku pacientů a následnou retrospektivní klinicko‑patologickou korelací analyzovat vliv jednotlivých komorbidit na celkový průběh onemocnění. Výsledná analýza totiž může mít u budoucích pacientů potenciální terapeutické konsekvence a být přínosem pro zpřesnění prognostických aspektů onemocnění.
Podpořeno granty Univerzity Karlovy PRVOUK-P26/LF1/4 a IGA MZ ČR: NT 12094-5/2011, NT13543-4/2012 a NT14145-3/2013.
Seznam použitých zkratek
AGD – demence s argyrofilními zrny
AN – Alzheimerova nemoc
DLB – demence s Lewyho tělísky
FTD – frontotemporální demence
FTLD – frontotemporální lobární degenerace
FTLD-MND – frontotemporální lobární degenerace s onemocněním motorického neuronu
MSA – multisystémová atrofie
NIA-AA – National Institute on Aging-Alzheimer’s Association
PART – věkově vázaná primární tauopatie
PSP – progresivní supranukleární obrna
TDP-43 – transactive response DNA binding protein 43
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Přijato k recenzi: 6. 5. 2015
Přijato do tisku: 29. 9. 2015
Podpořeno garantem: Institucionální podpory č. 2 RVO-FNOs/2013.
doc. MUDr. Robert Rusina, Ph.D.
Neurologická klinika a Centrum klinických neurověd
1. LF UK a VFN v Praze
Kateřinská 30
120 00 Praha 2
e-mail: robert.rusina@lf1.cuni.cz
Sources
1. Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC et al. National Institute on Aging ‑ Alzheimer‘s Association guidelines for the neuropathologic assessment of Alzheimer‘s disease. Alzheimers Dement 2012; 8(1): 1 – 13. doi: 10.1016/ j.jalz.2011.10.007.
2. Whitwell JL, Dickson DW, Murray ME, Weigand SD, Tosakulwong N, Senjem ML et al. Neuroimaging correlates of pathologically defined subtypes of Alzheimer‘s disease: a case ‑ control study. Lancet Neurol 2012; 11(10): 868 – 877. doi: 10.1016/ S1474 ‑ 4422(12)70200 ‑ 4.
3. Murray ME, Graff ‑ Radford NR, Ross OA, Petersen RC, Duara R, Dickson DW. Neuropathologically defined subtypes of Alzheimer‘s disease with distinct clinical characteristics: a retrospective study. Lancet Neurol 2011; 10(9): 785 – 796. doi: 10.1016/ S1474 ‑ 4422(11)70156 ‑ 9.
4. Mensikova K, Kanovsky P, Kaiserova M, Nestrasil I, Bares M. Proměnlivá tvář parkinsonské neurodegenerace. Cesk Slov Neurol N 2013; 76/ 109(1): 26 – 34.
5. Ahmed Z, Bigio EH, Budka H, Dickson DW, Ferrer I, Ghetti B et al. Globular glial tauopathies (GGT): consensus recommendations. Acta Neuropathol 2013; 126(4): 537 – 544. doi: 10.1007/ s00401 ‑ 013 ‑ 1171 ‑ 0.
6. Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF,Abner EL, Alafuzoff I et al. Primary age‑related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 2014; 128(6): 755 – 766. doi: 10.1007/ s00401 ‑ 014 ‑ 1349 ‑ 0.
7. Rohan Z, Matej R. Current concepts in the classification and diagnosis of frontotemporal lobar degenerations: a practical approach. Arch Pathol Lab Med 2014; 138(1): 132 – 138. doi: 10.5858/ arpa.2012 ‑ 0510 ‑ RS.
8. Bigio EH. TDP ‑ 43 variants of frontotemporal lobar degeneration. J Mol Neurosci 2011; 45(3): 390 – 401. doi: 10.1007/ s12031 ‑ 011 ‑ 9545 ‑ z.
9. Sutovsky S, Kralova M, Kollar B, Siarnik P, Dragasek J, Izakova L et al. Frontotemporálna lobárna degenerácia z pohľadu nových klinicko‑patologických korelácií. Cesk Slov Neurol N 2013; 76/ 109(6): 679 – 689.
10. Arnold SJ, Dugger BN, Beach TG. TDP ‑ 43 deposition in prospectively followed, cognitively normal elderly individuals: correlation with argyrophilic grains but not other concomitant pathologies. Acta Neuropathol 2013; 126(1): 51 – 57. doi: 10.1007/ s00401 ‑ 013 ‑ 1110 ‑ 0.
11. Rohan Z, Parobkova E, Johanidesova S, Koukolik F, Rusina R, Matej R. Lidské prionové nemoci v České republice – 10 let zkušeností s diagnostikou. Cesk Slov Neurol N 2013; 76/ 109(3): 300 – 306.
12. Dugger BN, Adler CH, Shill HA, Caviness J, Jacobson S, Driver ‑ Dunckley E et al. Concomitant pathologies among a spectrum of parkinsonian disorders. Parkinsonism Relat Disord 2014; 20(5): 525 – 529. doi: 10.1016/ j.parkreldis.2014.02.012.
13. Kovacs GG, Milenkovic I, Wohrer A, Hoftberger R, Gelpi E, Haberler C et al. Non ‑ Alzheimer neurodegenerative pathologies and their combinations are more frequent than commonly believed in the elderly brain: a community‑based autopsy series. Acta Neuropathol 2013; 126(3): 365 – 384. doi: 10.1007/ s00401 ‑ 013 ‑ 1157 ‑ y.
14. Rahimi J, Kovacs GG. Prevalence of mixed pathologies in the aging brain. Alzheimers Res Ther 2014; 6(9): 82. doi: 10.1186/ s13195 ‑ 014 ‑ 0082 ‑ 1.
15. Rusina R, Bourdain F, Matěj R. Atrophie multi‑systématisée et maladie d’Alzheimer: association rare de deux affections neuro‑dégénératives. Ŕ propos d’un cas. Revue Neurologique 2007; 163(12): 1239 – 1241.
16. Colom ‑ Cadena M, Gelpi E, Charif S, Belbin O, Blesa R, Marti MJ et al. Confluence of alpha ‑ synuclein, tau, and beta‑amyloid pathologies in dementia with Lewy bodies. J Neuropathol Exp Neurol 2013; 72(12): 1203 – 1212. doi: 10.1097/ NEN.0000000000000018.
17. Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B et al. Distinct alpha ‑ synuclein strains differentially promote tau inclusions in neurons. Cell 2013; 154(1): 103 – 117. doi: 10.1016/ j.cell.2013.05.057.
18. Frankova V, Matej R, Rusina R. Progredující demence s parkinsonizmem a poruchami chování – od prvních příznaků k neuropatologické diagnóze (kazuistika). Cesk Slov Neurol N 2015; 78(2): 209 – 214. doi: 10.14735/ amcsnn2015209.
19. Rusina R, Pazdera L, Kulistak P, Vysata O, Matej R. Pick and Alzheimer diseases: a rare comorbidity presenting as corticobasal syndrome. Cogn Behav Neurol 2013; 26(4): 189 – 194. doi: 10.1097/ WNn.0000000000000011.
20. Mensikova K, Matej R, Tuckova L, Rusina R, Ehrmann J, Kanovsky P. Progressive supranuclear palsy phenotype mimicking synucleinopathies. J Neurol Sci 2013; 329(1 – 2): 34 – 37. doi: 10.1016/ j.jns.2013.03.008.
21. Josephs KA, Whitwell JL, Parisi JE, Knopman DS, Boeve BF, Geda YE et al. Argyrophilic grains: a distinct disease or an additive pathology? Neurobiol Aging 2008; 29(4): 566 – 573.
22. Thal DR, Schultz C, Botez G, Del Tredici K, Mrak RE, Griffin WS et al. The impact of argyrophilic grain disease on the development of dementia and its relationship to concurrent Alzheimer‘s disease‑related pathology. Neuropathol Appl Neurobiol 2005; 31(3): 270 – 279.
23. Matej R, Koukolik F. Nemoc s argyrofilními zrny: kazuistické sdělení prvních dvou případů diagnostikovaných v ČR a přehled literatury. Ces Slov Patol 2006; 42(2): 66 – 70.
24. Josephs KA, Murray ME, Whitwell JL, Parisi JE, Petrucelli L, Jack CR et al. Staging TDP ‑ 43 pathology in Alzheimer‘s disease. Acta Neuropathol 2014; 127(3): 441 – 450. doi: 10.1007/ s00401 ‑ 013 ‑ 1211 ‑ 9.
25. Josephs KA, Whitwell JL, Weigand SD, Murray ME, Tosakulwong N, Liesinger AM et al. TDP ‑ 43 is a key player in the clinical features associated with Alzheimer‘s disease. Acta Neuropathol 2014; 127(6): 811 – 824. doi: 10.1007/ s00401 ‑ 014 ‑ 1269 ‑ z.
26. Jung Y, Dickson DW, Murray ME, Whitwell JL, Knopman DS, Boeve BF et al. TDP ‑ 43 in Alzheimer‘s disease is not associated with clinical FTLD or Parkinsonism. J Neurol 2014; 261(7): 1344 – 1348. doi: 10.1007/ s00415 ‑ 014 ‑ 7352 ‑ 5.
27. Rusina R, Sheardova K, Rektorova I, Ridzon P, Kulistak P, Matej R. Amyotrophic lateral sclerosis and Alzheimer‘s disease – clinical and neuropathological considerations in two cases. Eur J Neurol 2007; 14(7): 815 – 818.
28. Kovacs GG, Seguin J, Quadrio I, Hoftberger R, Kapas I, Streichenberger N et al. Genetic Creutzfeldt ‑ Jakob disease associated with the E200K mutation: characterization of a complex proteinopathy. Acta Neuropathol 2011; 121(1): 39 – 57. doi: 10.1007/ s00401 ‑ 010 ‑ 0713 ‑ y.
29. Vital A, Canron MH, Gil R, Hauw JJ, Vital C. A sporadic case of Creutzfeldt ‑ Jakob disease with beta‑amyloid deposits and alpha ‑ synuclein inclusions. Neuropathology 2007; 27(3): 273 – 277.
30. Rodriguez ‑ Diehl R, Rey MJ, Gironell A, Martinez ‑ Saez E, Ferrer I, Sanchez ‑ Valle R et al. „Preclinical“ MSA in definite Creutzfeldt ‑ Jakob disease. Neuropathology 2012; 32(2): 158 – 163. doi: 10.1111/ j.1440 ‑ 1789.2011.01232.x.
31. Castellani RJ, Perry G, Smith MA. Prion disease and Alzheimer‘s disease: pathogenic overlap. Acta Neurobiol Exp (Wars) 2004; 64(1): 11 – 17.
32. Alafuzoff I, Gelpi E, Al ‑ Sarraj S, Arzberger T, Attems J, Bodi I et al. The need to unify neuropathological assess-ments of vascular alterations in the ageing brain: multicentre survey by the Brainnet Europe consortium. Exp Gerontol 2012; 47(11): 825 – 833.
33. Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW et al. National Institute on Aging ‑ Alzheimer‘s Association guidelines for the neuropathologic assessment of Alzheimer‘s disease: a practical approach. Acta Neuropathol 2012; 123(1): 1 – 11. doi: 10.1007/ s00401 ‑ 011 ‑ 0910 ‑ 3.
34. Rusina R, Matěj R et al. Neurodegenarativní onemocnění. Praha: Mladá fronta 2014.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2015 Issue 6
Most read in this issue
- Nádory očnice
- Novinky ze světa NOAK – „Dienerovo pravidlo 1- 3- 6- 12“ a první antidotum s potvrzeným účinkem
- Psychometrické vlastnosti české verze Epworthské škály spavosti
- Familiární amyloidová polyneuropatie – kazuistika