
Příčiny vzniku Parkinsonovy nemoci – nové představy a nové výzvy
Etiology of Parkinson’s Disease – New Advances and New Challenges
Etiology of Parkinson’s disease (PD) for many years has been remaining the matter of active discussions and intensive interdisciplinary studies. About 5– 10% of all cases of PD are represented by monogenic forms manifesting predominantly in younger persons, while most cases of the disease are sporadic and have multifactorial nature. The key molecular event in the development of neurodegeneration in PD is the conformational change of a small vesicular protein α‑ synuclein initiating its self‑ fibrilization with forming neurotoxic cytoplasmic aggregates and Lewy bodies/ neurites. Misfolding of α‑ synuclein in PD is caused by specific interaction of environmental factors, genomic factors and characteristics of systemic metabolism, which, in combination, determines the processes of cell detoxication, mitochondrial functioning, synaptic transmission and endosomal transport. In this review, possible exogenous and endogenous triggers of pathological process in PD are discussed in detail. Among the risk factors, special consideration is given to the role of different neurotoxins, prion hypothesis of the development of PD, as well as up‑ to‑ date knowledge about genetics of familial and sporadic cases of PD.
Key words:
Parkinson’s disease – etiology – molecular mechanisms – risk factors – genetics
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Authors:
S. N. Illarioškin
Authors‘ workplace:
Výzkumné centrum neurologie, Ruská akademie lékařských věd, Moskva
Published in:
Cesk Slov Neurol N 2015; 78/111(3): 283-291
Category:
Review Article
Overview
Příčiny vzniku Parkinsonovy nemoci (PN) zůstávají po mnoho let předmětem živých diskuzí a intenzivních mezioborových výzkumů. Přibližně 5– 10 % všech případů PN představují monogenní formy, které se projevují především u osob mladšího věku, zatímco většinu případů tvoří sporadická onemocnění multifaktoriálního rázu. Klíčovou molekulární událostí v rozvoji neurodegenerace při PN je porucha konformace malé vezikulární bílkoviny α‑ synukleinu, jež iniciuje její fibrilizaci s tvorbou neurotoxických cytoplazmatických agregátů, Lewyho tělísek a Lewyho neuritů. Patologie α‑ synukleinu při PN je podmíněna specifickým vzájemným působením faktorů prostředí, zvláštností genomu a systémového metabolizmu, což v souhrnu určuje charakter procesů buněčné detoxikace, fungování mitochondrií, synaptické transmise a endozomálního transportu. V přehledu jsou podrobně posouzeny možné exogenní a endogenní spouštěče patologického procesu při PN. Zvláštní pozornost při analýze rizikových faktorů náleží úloze různých neurotoxinů, prionové hypotéze rozvoje PN a rovněž současným představám o genetice familiárních a sporadických případů PN.
Klíčová slova:
Parkinsonova nemoc – etiologie – molekulární mechanizmy – rizikové faktory –genetika
Úvod
Vývoj pohledů na příčiny Parkinsonovy nemoci (PN) v průběhu téměř 200 let odrážel úroveň rozvoje neurologie v tom či jiném časovém období. Prošel složitou cestu od uznání výlučně exogenní povahy nemoci až po absolutizaci úlohy genetiky v její etiologii. Ukázalo se, že pravda je někde uprostřed: dnes je pevně stanoven význam jak exogenních, tak endogenních mechanizmů vzniku PN, přičemž v různých věkových skupinách se poměr těchto faktorů liší. Přibližně 5– 10 % všech případů PN představují dědičné (monogenní) formy, které se projevují především u osob mladšího věku [1– 3], zatímco absolutní většina případů je sporadických a mají multifaktoriální ráz.
Podle současných představ vedoucí úloha v rozvoji neurodegenerativního procesu náleží malé presynaptické bílkovině α‑ synukleinu. V normálních podmínkách se α‑ synuklein v buňce nachází ve formě tetrameru [4] a pravděpodobně se účastní procesů vezikulárního transportu a regulace dopaminergního přenosu [5– 7]. Tato bílkovina se široce exprimuje v různých oddílech centrálního nervového systému (CNS) a tvoří cca 1 % celkové hmotnosti mozku, avšak její přesné fyziologické funkce zatím neznáme. Klíčovou etapou molekulární patogeneze PN je změna nativní prostorové konformace α‑ synukleinu se vznikem složitějších β‑ struktur a neurotoxických oligomerů, jejich další fibrilizace a tvorba rostoucích cytoplazmatických agregátů [8– 12]. Právě α‑ synuklein je základní komponentou Lewyho tělísek a Lewyho neuritů – klasických morfologických ukazatelů PN [13].
U dědičných forem PN je porucha konformace α‑ synukleinu a jeho agregace v neuronech podmíněna mutacemi velkého počtu genů „synukleinové kaskády“ (viz dále). Při častější sporadické formě PN souvisí patologie α‑ synukleinu se specifickým vzájemným působením exogenních vlivů, zvláštností genomu a systémového metabolizmu. Tyto faktory se přidávají k věkovému faktoru a celkově určují charakter procesů buněčné detoxikace, fungování mitochondrií, synaptické transmise a endozomálního transportu u konkrétního jedince [14– 19]. Jednou z výzev, která dnes stojí před neurovědou, je stanovení klíčových molekulárních událostí iniciujících poruchy konformace α‑ synukleinu a další posloupnosti biochemických reakcí, neboť právě toto může pomoci při vypracování nových přístupů k rané terapii PN.
Faktory prostředí
Neurotoxiny
Z exogenních faktorů PN se dnes může za nejlépe prokázanou považovat etiopatogenetická role řady neurotoxinů, zejména pesticidů [20,21]. Ukázalo se, že chronické systémové působení rotenonu, paraquatu, epoxomicinu a dalších podobných sloučenin v experimentu vyvolává mnohé klinické, neurochemické a patomorfologické charakteristiky PN [22– 24]. Epidemiologické výzkumy tyto závěry potvrzují. Četné studie ukázaly zvýšené riziko PN (průměrně 1,4krát) ve spojení s farmářstvím a bydlením v zemědělských agroprůmyslových oblastech – to jest s faktory, které předpokládají mnohaletý kontakt s pesticidy [18,25– 28]. Bylo prokázáno synergické působení různých pesticidů ve vztahu k PN: například současný chronický kontakt s manebem a paraquatem zvyšuje riziko vzniku PN o 75 % [29]. Nebezpečí stoupá při prodlouženém kontaktu s pesticidy, a naopak při používání jednoduchých ochranných prostředků (rukavice, smývání toxinu vodou, když se dostane do kontaktu s pokožkou atd.) se snižuje [21,27,30].
Důležitým potvrzením souvislosti pesticidů s PN byly údaje ze zkoumání mozků pacientů post mortem. Ukázalo se, že hladina insekticidu dieldrin (ve vyspělých zemích platí zákaz jeho používání již od 70. let 20. století) v nucleus caudatus pacientů s PN je významně vyšší než v kontrolních mozcích [31]. Dieldrin byl zjištěn v šesti ze 20 vzorků mozku pacientů s PN a ani v jednom ze 14 kontrolních vzorků [32].
Pozitivní asociace mezi expozicí pesticidům a rizikem vzniku PN je tedy zcela zjevná. Molekulárním základem uvedené asociace je skutečnost, že pesticidy (rotenon, paraquat, maneb a další) mohou vyvolávat konformační změny molekuly buněčné bílkoviny α‑ synukleinu a podstatně zrychlovat tempo tvorby α‑ synukleinových fibril a Lewyho tělísek v neuronech [33,34]. Důležitý význam při vzniku parkinsonizmu se přikládá rovněž schopnosti pesticidů narušovat fungování mitochondrií (inhibicí komplexu I. dýchacího řetězce), vyvolávat oxidační stres a apoptotické reakce, snižovat aktivitu ubikvitin‑proteazomového systému a též řadě dalších vlastností těchto sloučenin (přehled viz v [35]).
Parkinsonský syndrom vyvolávají rovněž neurotoxiny, které specificky ničí buňky středního mozku produkující dopamin – 6- hydroxydopamin a N‑ metyl‑ 4- fenyl 1,2,3,6- tetrahydropyridin (metylfenyltetrahydropyridin; MPTP) [23,36,37]. Interagují s dopaminovými transportéry, výběrově se akumulují v nigrostriatálních neuronech a vyvolávají jejich zánik v důsledku monoaminooxidázou‑ B (МАО‑ В) zprostředkované přeměny na toxické metabolity rázu volných radikálů. Předpokládá se, že tyto sloučeniny se mohou dostávat do organizmu s potravou, vodou, vzduchem. Působení dalších, v literatuře předpokládaných parkinsonských neurotoxinů (takových jako mangan, železo, metanol a další) zřejmě rovněž může přispívat k rozvoji PN pomocí prozkoumaných mechanizmů – agregace α‑ synukleinu, poškození mitochondrií, indukce oxidačního stresu, inhibice proteazomové aktivity [21,27,38– 40].
Úlohu exogenních toxických sloučenin při vzniku PN podporují současné představy o nejranějších stadiích neurodegenerativního procesu u tohoto onemocnění. Bylo zjištěno, že ještě před prvními projevy pohybových poruch typických pro PN se patologické synuklein‑pozitivní inkluze objevují v čichových bulbech a v neuronech visceromotorického jádra vagu (nucleus dorsalis nervi vagi) a poté se postupně šíří rostrálně (do jader nuclei raphe a retikulární formace, locus coeruleus, substantia nigra atd.) [41,42]. Kromě toho se v latentním období PN zřetelné neurodegenerativní změny parkinsonského typu (agregáty α‑ synukleinu, Lewyho tělíska a Lewyho neurity) objevují v periferních vegetativních neuronech: v buňkách Meissnerova a Auerbachova plexu střevního traktu, v prevertebrálních a paravertebrálních gangliích, v distálních sympatických zakončeních a v gangliích truncus sympathicus, v neuronech nadledvinek, prostaty, slinných žláz, kůže atd. [43– 46]. Ukázalo se, že některé toxiny ze zevního prostředí napomáhají uvolňování α‑ synukleinu enterickými neurony; bílkovina je poté zachycena presynaptickými vegetativními zakončeními a retrográdně se transportuje do těla neuronu, což je podkladem progrese parkinsonské patologie [47]. Primární kultury enterických neuronů mohou vylučovat α‑ synuklein cestou klasické exocytózy a tato sekrece je regulována velice složitými mechanizmy [48]. Máme tedy důvody považovat postižení vegetativních neuronů střevního traktu a buněk čichových jader za nejranější děje v průběhu PN, které ukazují na možnost vdechnutí a/ nebo alimentárního příjmu spoušťového patogenu s následným rozšířením patologického procesu po vláknech čichového nervu a n. vagus [49].
Význam exogenních činitelů při realizaci predispozice k PN podtrhují nálezy různých biologicky aktivních látek, které mohou riziko vzniku PN snižovat. K takovým dokázaným nebo předpokládaným faktorům zevního prostředí snižujícím riziko PN patří: kouření tabáku, pití kávy a možná i některých druhů čaje, užívání blokátorů kalciových kanálů, nesteroidních antiflogistik, velkých dávek vitaminu Е a další [20, 21,50– 52]. Přitom může být ochranné působení uvedených faktorů prostředí zprostředkováno rovněž jejich vlivem na molekulární vlastnosti α‑ synukleinu. Je například dokázáno, že komponenty tabákového kouře nikotin a hydrochinon stabilizují rozpustné oligomerní formy α‑ synukleinu a inhibují tvorbu synukleinových fibril [53].
Zkoumání celé patofyziologické palety složitého vzájemného působení potenciálních neurotoxinů a neuroprotektorů, jež určují riziko poškození monoaminergních neuronů CNS, je v současné době jedním z nejperspektivnějších směrů při vývoji nových metod léčby PN.
Priony a prionová hypotéza
Nezávisle na tom, z jakého důvodu vzniká patologická konformace α‑ synukleinu (např. pod vlivem výše uvedených toxických agens nebo v důsledku náhodných událostí ve stárnoucích neuronech), má další dynamika α‑ synukleinové patologie mnoho společného s mechanizmy prionových onemocnění [54,55]. Patologická forma α‑ synukleinu může vystupovat v buňce jako matrice, na níž probíhá autoreplikace anomálních molekul v interakci s okolními normálními molekulami α‑ synukleinu [56,57]. Dále dochází k postupnému růstu neuronálních bílkovinných agregátů, od malých oligomerů a protofibril až po masivní strukturované inkluze – Lewyho tělíska, jež do své struktury zabírají mnoho dalších buněčných bílkovin [10,12,58]. Podobnost s prionovými onemocněními je zdůrazněna amyloidním charakterem agregátů α‑ synukleinu tvořících se v cytoplazmě neuronů [59].
Předpoklad prionové povahy neurodegenerativního procesu s účastí α‑ synukleinu původně vznikl na základě výsledků morfologického výzkumu mozků pacientů s PN, kterým byly 15– 20 let před smrtí implantovány fetální mezencefalické neurony do corpus striatum. Ukázalo se, že těmto pacientům se v implantovaných neuronech tvořila Lewyho tělíska, což s veškerou jasností svědčí o předání patologického procesu z „nemocných“ neuronů pacienta intaktním buňkám zavedeným zvenku [60,61]. Možnost předávání α‑ synukleinové patologie od neuronu k neuronu byla poté demonstrována rovněž na transgenních živočiších a buněčných kulturách [62– 64]. Vyšlo najevo, že α‑ synuklein mohou různé typy buněk CNS vylučovat a zachycovat za pomoci řady samostatných molekulárních mechanizmů [65] a patologické bílkoviny se objevují v mozkomíšním moku pacientů s PN [66].
Údaje o transsynaptickém předávání patologické formy α‑ synukleinu od buňky k buňce se dobře shodují s pozorováním, že lewyovská patologie při PN postupuje především podél vytvořených nervových drah [42,67].
Poslední tři roky poznamenala série působivých experimentálních prací, které potvrzují schopnost α‑ synukleinu iniciovat v mozku po inokulaci patologické formy bílkoviny proces podobný prionovému. Bylo zjištěno, že zavedení purifikovaných α‑ synukleinových fibril do corpus striatum vyvolává u myší degeneraci buněk substantia nigra, objevují se charakteristické fibrilární synukleinové agregáty a pohybové poruchy [68,69]. Synukleinové agregáty se přitom objevovaly i v dalších oddílech mozku v přísné posloupnosti, která odrážela nikoli blízkost k místu injekce, ale konektivitu CNS. To svědčí o rozšiřování patologie od prvotního místa zavedení patologické bílkoviny na značnou vzdálenost, po drahách vedoucích do CNS. Uvedený efekt nebyl pozorován u transgenních myší s potlačenou expresí vlastního α‑ synukleinu, což potvrzuje, že k šíření „parkinsonské“ neurodegenerace je nezbytná interakce patologického α‑ synukleinu s normálními molekulami [67].
Diseminace α‑ synukleinových agregátů v mozku probíhá rovněž po inokulaci patologického materiálu uvolňovaného z mozků transgenních živočichů nebo lidí s nejrůznějšími formami synukleopatie. Projevily se „infekční“ vlastnosti homogenátů z mozků transgenních myší exprimujících lidský gen SNCA s mutací A53T, jež jsou klasickým modelem PN: zavádění těchto homogenátů do corpus striatum a kůry myší stejné genetické linie značně urychlilo rozvoj synukleopatie v mozku a úhyn živočichů [70,71]. Analogickým způsobem nitromozková inokulace extraktů lidských Lewyho tělísek [69,72] nebo gliálních α‑ synukleinových inkluzí, charakteristických pro multisystémovou atrofii [73], vyvolávala u myší vznik a šíření cerebrálních agregátů α‑ synukleinu a vznik pohybových poruch. Stejný výsledek byl zjištěn u makaků, přičemž neurodegenerace po inokulaci extraktů α‑ synukleinu do mozku v průběhu 4– 17 měsíců pozorování neustále narůstala [72].
Zajímavé je, že Guo et al [74] prokázali existenci různých konformačních variant („strains“) α‑ synukleinu, které v experimentu in vivo vyvolávaly různé formy cerebrální patologie. Možná že to částečně vysvětluje význačnou klinickou a morfologickou heterogennost onemocnění, patřících do skupiny synukleopatií.
Analogicky jako u α‑ synukleinu byl v poslední době prokázán přenos i řady dalších „neprionových“ bílkovin – β‑ amyloidu, tau proteinu, TDP‑ 43, odpovědných za vznik Alzheimerovy choroby, nemocí motoneuronu a dalších neurodegenerativních patologií [75,76]. Avšak v nedávné práci Irwina et al se nepodařilo prokázat ani zvýšený výskyt onemocnění Parkinsonovou či Alzheimerovou chorobou ve velké skupině příjemců kadaverózního růstového hormonu, ani významné nahromadění patologických forem α‑ synukleinu, β‑ amyloidu nebo fosforylovaného tau proteinu v nekroptických tkáních hypofýzy pacientů s neurodegenerativními onemocněními [77]. Proto, na rozdíl od klasických prionových onemocnění, zatím nemáme údaje o přímém infekčním předávání PN nebo Alzheimerovy choroby přes kontaminovaný biologický materiál.
Hypotéza o prionové podstatě PN tedy vyžaduje další zkoumání a detailnější experimentální zdůvodnění. Nicméně již dnes tyto představy o vzniku PN umožňují navrhovat řadu nových molekulárních metod terapie. In vitro i in vivo se ukázalo, že podávání monoklonálních anti‑synukleinových protilátek odvrací přenos patologické bílkoviny od neuronu k neuronu tím, že zabrání zachycení „prionu podobného“ α‑ synukleinu okolními intaktními buňkami [78]. Používání takových protilátek u transgenních myší je doprovázeno sníženou tvorbou Lewyovské patologie v mozku, což možná otevírá novou cestu v léčbě PN.
Kraniocerebrální trauma
Dobře známá zvláštní forma sekundárního parkinsonizmu je projevem traumatické encefalopatie, jež vzniká buď v důsledku četných opakovaných otřesů mozku v rizikových skupinách („encephalopathia pugilistica“ aj.) nebo v důsledku těžkého (včetně jednorázového) traumatického poškození bazálních ganglií a/ nebo jejich spojů [79]. To se stalo podkladem velkého počtu epidemiologických studií věnovaných možné souvislosti kraniocerebrálního traumatu a idiopatické PN. Přibližně ve třetině všech studií byla u pacientů zjištěna statisticky významná asociace prodělaných kraniocerebrálních traumat různé závažnosti v anamnéze s rozvojem PN [80,81]. Podle údajů nedávné metaanalýzy zahrnující 22 publikovaných studií [82] i anamnestický výskyt jediného kraniocerebrálního traumatu doprovázeného poruchou vědomí se věrohodně asociuje se zvýšeným rizikem vzniku PN (OR 1,57; 95% CI; 1,35– 1,83). Je zajímavé, že i při nevýznamné asociaci byla prakticky ve všech sériích pozorování u pacientů s PN četnost událostí souvisejících s prodělaným kraniocerebrálním traumatem vyšší než v kontrolní skupině [80], což slouží jako nepřímý důkaz úlohy kraniocerebrálního traumatu při vytváření rizika vzniku PN. Má se za to, že toto zvýšené riziko může mít mechanické vysvětlení (rotační poškození vláken neuronů mezencefalu), kdy dochází k narušení hematoencefalické bariéry a k odpovídající imunitní odpovědi ze strany mozkové tkáně, s aktivováním reakce „akutní fáze“ s hyperexpresí bílkovin vyvolávajících vznik α‑ synukleinových agregátů v cytoplazmě neuronů [83]. Lze rovněž předpokládat, že při kraniocerebrálním traumatu dochází k prudkému zrychlení šíření patologie α‑ synukleinu do CNS v souvislosti s masivním poškozením axonů, již obsahujících počáteční subklinické projevy α‑ synukleinové patologie [83, 84].
Věkové a konstituční faktory
Věk je nejlépe prokázaným nezávislým faktorem rizika vzniku PN. Je dobře známo, že při průměrném populačním výskytu 120– 180 případů na 100 000 obyvatel, incidence PN u osob starších 60 let dosahuje 1 %, ale ve skupině osob starších 80 let je to již kolem 4 % [21,85]. Na světě se nachází celkově okolo šesti milionů nemocných s PN, z nichž velká část připadá na obyvatelstvo v pokročilém věku (pouze každý 10. pacient, jenž onemocněl PN, je mladší 50 let) [85]. Právě věkovým faktorem se odůvodňují prognózy WHO o tom, že do roku 2030 se očekává zdvojnásobení počtu pacientů s PN a v roce 2050 bude jejich počet již čtyřnásobný – a to v důsledku závislosti PN na věku a tendence k neustálému stárnutí obyvatelstva ve vyspělých zemích světa.
Příčiny spojení PN s pokročilým věkem byly dostatečně prozkoumány a souvisejí s vyčerpáním plasticity CNS v důsledku stárnutí [12,16,58,86,87]. Stav organizmu ve zralém a stařeckém věku se vyznačuje:
- chronickým oxidačním stresem,
- nahromaděním mutací mitochondriální DNA,
- snížením hladiny glutationu a dalších složek antioxidační ochrany,
- potlačením funkce ubikvitin‑proteazomového systému,
- snížením schopnosti neuronů aktivovat stresovou odpověď.
Jakýkoli z těchto faktorů nebo jejich kombinace přispívá k tomu, že počáteční změny ukládání α‑ synukleinu či jiných cílových neuronálních bílkovin, které by u mladých osob byly poměrně snadno odstraněny výkonnými endogenními ochrannými systémy, se u lidí pokročilého věku stávají neodvratnými a vedou ke „spuštění“ fatální cytotoxické kaskády a k úhynu buňky [8,16].
Zvýšené riziko vzniku PN se spojuje i s řadou dalších konstitučních faktorů [21,85]:
- a) mužským pohlavím (předpokládá se ochranné působení estrogenů u žen);
- b) rezavou barvou vlasů (načervenalý odstín vzniká při nerovnováze pigmentů – nadbytku feomelaninu a nedostatku zrnitého melaninu, který má přímý vztah k biochemii nigrálních neuronů);
- c) nadbytečnou tělesnou hmotností (možná je tento faktor druhotný ve vztahu k tělesné aktivitě, je však známo, že vysoká fyzická aktivita snižuje riziko PN).
Genetické faktory a jejich interakce s faktory vnějšího prostředí
Úlohu dědičnosti při vzniku PN podrobně popsalo mnoho epidemiologických a populačních studií [1,21,86,88]. Analýzou rozsáhlých souborů pacientů bylo prokázáno, že pozitivní rodinná anamnéza je jedním z vedoucích rizikových faktorů vzniku PN. U parkinsonizmu existuje výrazná tendence ke hromadění případů onemocnění uvnitř rodiny a pozitivní rodinná anamnéza se nachází u 10– 24 % nemocných. Riziko vzniku nemoci u příbuzných 1. stupně se pohybuje v rozmezí 4– 10 % a značně převyšuje riziko v celé populaci. Rodinný výskyt je zvlášť charakteristický pro rané (do 40 let) případy PN. Výzkumy párů dvojčat s PN, kde byl hodnocen metabolizmus dopaminu v bazálních gangliích pomocí PET, rovněž ukázaly vysokou konkordanci monozygotních dvojčat (55 %) ve srovnání s dizygotními (18 %), což vypovídá o příspěvku genetiky ke vzniku PN [89].
Přímým potvrzením významu genetických faktorů při vzniku PN jsou rodiny, v nichž se popisuje dědičnost onemocnění podle Mendelových zákonů (nejčastěji autozomálně dominantního a autozomálně recesivního typu). Zejména v těchto familiárních případech PN byly identifikovány geny a odpovídající bílkovinné produkty, které umožnily odhalit klíčové vazby patobiochemické kaskády u PN [2,3,12,15,90].
Dnes jsou známy genetické lokusy 20 dědičných monogenních forem PN (tab. 1), což dosvědčuje výraznou genetickou heterogenitu primárního parkinsonizmu [3,15,91]. Největší význam má šest níže uvedených monogenních forem PN.
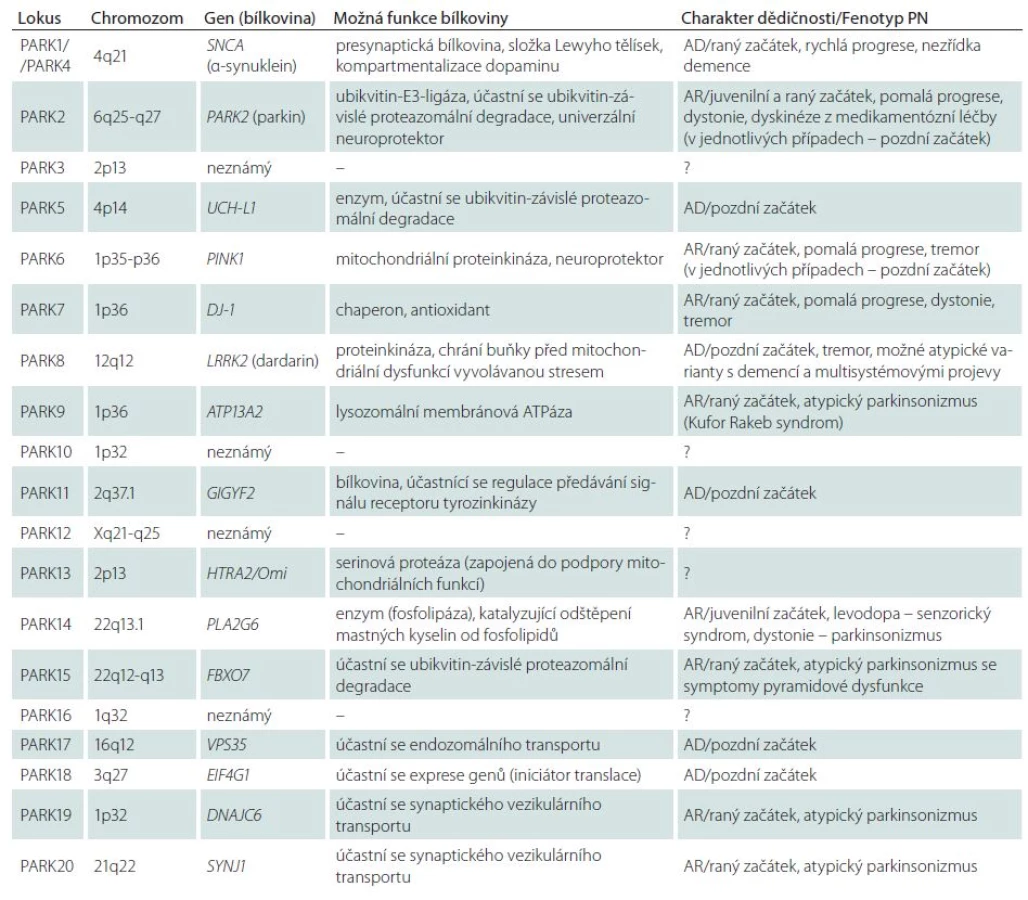
PARK1 – autozomálně dominantní forma PN, je podmíněna mutacemi v genu pro α‑ synuklein (SNCA). V rodinách z různých populací světa je dnes popsáno šest bodových mutací v genu pro α‑ synuklein, avšak nejčastěji se při PARK1 formě PN setkáváme s úplnou multiplikací (obvykle duplikace) celého lokusu s genem SNCA v chromozomální oblasti 4q21. Bylo spočítáno, že s duplikací tohoto chromozomového lokusu se setkáváme přibližně v 1– 2 % všech případů PN s autozomálně dominantní dědičností. Je zajímavé, že v případech triplikace lokusu 4q21 může mít klinický obraz závažnější charakter odpovídající demenci s Lewyho tělísky [92], což demonstruje význam dávky genu SNCA při tvorbě klinického i morfologického fenotypu.
PARK2 – autozomálně recesivní forma PN, je podmíněna mutacemi ve stejnojmenném genu, kódujícím bílkovinu parkin. Mutace genu PARK2 jsou nejčastější příčinou raného (včetně juvenilního) autozomálně recesivního parkinsonizmu: podmiňují přibližně 50 % familiárních a 18 % sporadických případů primárního juvenilního parkinsonizmu s počátkem do 20 let. Nedávný přehled velkého počtu studií, zahrnujících 3 952 pacientů různé etnické příslušnosti ukázal, že mutace PARK2 podmiňují 15,5 % familiárních a 4,3 % sporadických případů PN s raným počátkem symptomů [93]. Objevil se předpoklad, že nositelství mutací PARK2 dokonce i v heterozygotním stavu může být významným rizikovým faktorem vzniku PN [94]. Bílkovina parkin představuje Е3 ubikvitin ligázu, jejíž funkce spočívá v přenosu anomálních substrátů do proteazomového komplexu buňky ke štěpení. Velký význam se přikládá úloze parkinu při odstraňování poškozených mitochondrií z buňky prostřednictvím specializované různorodosti autofagie označované jako „mitofagie“ [95,96]. V současnosti se parkin bere jako polyvalentní neuroprotektivní agens mající klíčový význam pro přežití dopaminergních neuronů při působení různých neurotoxinů.
PARK6 – další autozomálně recesivní forma PN, je podmíněna mutacemi genu PINK1 (PTEN‑induced putative kinase 1). S patogenními mutacemi PINK1 se setkáváme v raném familiárním parkinsonizmu 20krát častěji u pacientů z asijských populací než u představitelů bílé rasy (13,5 % a 0,6 %) [93]. Mutace v daném genu se objevují rovněž v 1– 4 % sporadických případů PN s raným počátkem symptomů [88]. Stejně jako u formy PARK2 je popsán značný počet familiárních a sporadických případů PN asociovaných s heterozygotními mutacemi v genu PINK1. Současné údaje svědčí o tom, že bílkovina PINK1 spolupůsobí s parkinem v procesu označování defektních mitochondrií a zajišťování jejich degradace pomocí mitofagie [95]. To potvrzuje úlohu mitochondriální dysfunkce v molekulární patogenezi PN.
PARK7 – autozomálně recesivní forma, klinicky shodná s PARK2 a PARK6, je podmíněna mutacemi genu DJ‑ 1. Podíl pacientů s mutacemi daného genu mezi všemi případy autozomálně recesivního parkinsonizmu s raným počátkem symptomů činí pouze 0,4 % [93]. Bez ohledu na vzácnost této formy její objev umožnil doložit význam oxidačního stresu v patogenezi PN, jelikož bílkovina DJ‑ 1 je mitochondriální antioxidant, který neutralizuje přebytek H2O2 [97]. Zajímavé je, že DJ‑ 1 spolupůsobí s parkinem a PINK1 při regulaci mitochondriální dynamiky [98].
PARK8 – autozomálně dominantní forma, je podmíněna mutacemi v genu LRRK2. Gen LRRK2 (Leucine‑ Rich Repeat Kinase 2) má mimořádně velký význam při vzniku PN v celkové populaci. Bylo prokázáno, že mutace v něm podmiňují 1−7 % všech sporadických a familiárních případů PN v evropských populacích a 20−40 % případů v některých populacích regionů Blízkého východu a Středomoří. Celkově jsou mutace LRRK2 v současnosti posuzovány jako nejčastější genetická příčina familiární PN na celém světě (~7 %) [99,100]. Asi 4– 5 % familiárních a okolo 1– 2 % sporadických případů PN v evropských populacích souvisí s mutací G2019S, kdežto v Asii je nejčastější varianta G2385R. Bílkovinný produkt genu, dardarin, je cytoplazmatická GTP‑ vázaná kináza, pravděpodobně zapojená do zpracování neuronálních bílkovin a fungování mitochondrií. V mechanizmech neurotoxického působení LRRK2- asociovaných mutací se základní význam přikládá patologickému zvýšení kinázové aktivity enzymu dardarinu. V souvislosti s tím je identifikace molekulárních substrátů LRRK2 v současné době velmi aktuální problém neurologie.
PARK17 – první genetická forma PN, jejíž gen VPS35 (Vacuolar Protein Sorting 35) byl objeven za pomoci nové technologie exomového sekvenování [101,102]. Jak ukázaly následné studie, s touto formou PN se setkáváme v různých populacích, jsou popsány jak autozomálně dominantní, tak i sporadické případy [103]. Bílkovina VPS35 je složkou složitého komplexu asociovaného s cytozolovým povrchem endozomů a zprostředkujícího retrográdní transport mezi endozomy a Golgiho retikulem [88]. V souvislosti s tím lze předpokládat, že mutace genu VPS35 jsou doprovázeny poruchami endozomálně lysozomálního transportu a neuronální homeostázy.
Ostatní genetické formy PN zůstávají ojedinělé nebo jsou dosud málo prozkoumané a jejich místo v celkovém spektru primárního parkinsonizmu v různých populacích světa nebylo s konečnou platností stanoveno. Například formy PARK5 a PARK11 jsou popsány pouze v ojedinělých rodinách a údaje z originálních publikací tak nebyly potvrzeny dalšími autory. Formy PARK9, PARK14, PARK15, PARK19 a PARK20 jsou charakterizovány zvláštními fenotypy – kombinací parkinsonizmu s dystonií, spasticitou, kognitivními poruchami, supranukleární pohledovou obrnou, epileptickými záchvaty (syndrom Kufor‑ Rakeb, palido‑ pyramidový syndrom atd.). Je zřejmé, že tyto vzácné autozomálně recesivní syndromy mají s klasickou PN málo společného. Určité otázky zůstávají i u dalších forem PN, které jsou uvedeny v tab. 1.
Genetika jistým způsobem přispívá i ke vzniku sporadických případů PN, a to vytvářením predispozice k tomuto onemocnění [3]. Nejprozkoumanější a nejpřesvědčivější je v genezi sporadické PN úloha genu SNCA (α‑ synuklein). Bylo prokázáno, že dlouhé alely, související s dinukleotidovým polymorfizmem NACP‑ Rep1 v promotorové oblasti SNCA, zvyšují riziko PN přibližně 1,4krát v důsledku zvýšení exprese genu, což je provázeno zvýšením koncentrace α‑ synukleinu a jeho shlukováním v buňce [104– 106]. Ukázala se silná asociace sporadické PN s řadou dalších polymorfizmů a haplotypů v lokusu SNCA [106,107]. Souvislost variability SNCA s predispozicí k PN je zdůrazněna nedávným objevem zvláštního transkriptu α‑ synukleinu s prodlouženou 3’- netranslační oblastí – aSynL v mozcích pacientů [108]. Zjistilo se, že pro PN je charakteristické zvýšení poměru aSynL k celkovému α‑ synukleinu, přičemž přítomnost aSynL v buňce je nejdůležitějším faktorem patologické akumulace α‑ synukleinu a poruchy jeho buněčného rozložení. Je zajímavé, že poměr aSynL/ celkový α‑ synuklein se zvětšuje působením faktorů zevního prostředí souvisejících s oxidačním stresem a s rizikem vzniku PN (rotenon, МРТР) a naopak zmenšuje působením nikotinu, známého faktoru snižujícího riziko PN.
Patologie α‑ synukleinu zvyšující jeho sklon k agregaci s tvorbou fibrilárních struktur má tedy univerzální význam v patogenezi familiárních a sporadických případů PN. Tyto změny probíhají pod vlivem dědičných mutací, genetických polymorfizmů nebo různých výše uvedených exogenních neurotoxických faktorů.
Známé jsou i další kandidátní geny, které vymezují riziko vzniku sporadické PN, avšak predispoziční role každého konkrétního genu je relativně malá. K prokázaným genetickým rizikovým faktorům sporadické PN, kromě SNCA, patří heterozygotní mutace v genu GBA, polymorfizmy a haplotypy v genech LRRK2 a MAPT a různé polymorfní varianty v lokusu PARK16 [3,88]. Zvláštní pozornost si zaslouží gen GBA kódující lysozomální enzym glukocerebrosidázu. Homozygotní mutace v daném genu vyvolává vznik dobře známého lysozomálního onemocnění, Gaucherovy choroby, která se projevuje multiorgánovým postižením včetně parkinsonizmu a dalších neurologických projevů. Ukázalo se, že heterozygotní nosičství mutací v genu GBA je spojeno s pětinásobným zvýšením rizika PN a určuje vysokou pravděpodobnost vzniku kognitivních poruch u PN [109,110]. V souvislosti s tím se gen GBA dnes považuje za jeden z hlavních faktorů vzniku primárního parkinsonizmu v populaci.
V posledních letech byly značně rozvinuty technologie celogenomového skenování (Genome‑ Wide Association Studies; GWAS), které umožňují provádění úplného skríningu variabilních částí genomu za účelem vyhledávání možných asociací s těmi či jinými onemocněními. Nedávné výsledky celogenomového skenování u sporadické PN svědčí o existenci více než 10 nových genových lokusů asociovaných s tímto onemocněním (HLA‑DR, BST1, GAK/ DGKQ, MCCC1/ LAMP3, SYT11/ RAB25, FGF20, ACMSD, STK39, RAB7L1 a další) [111– 115]. Pozornost přitahuje asociace PN s geny imunitní odpovědi HLA‑DR. Tento nález potvrzuje úlohu zánětlivých reakcí mikroglie v patogenezi PN, což bylo zjištěno v řadě morfologických a neurozobrazovacích studií [116,117]. I další parkinsonské mutace mohou přispívat k aktivaci mikroglie [118]. Předpokládá se, že mikrogliální zánět zprostředkovává svůj efekt přes sekreci specifických cytokinů zesilujících agregaci α‑ synukleinu.
Genetický profil predispozice k PN stanovený pomocí GWAS má určité populační zvláštnosti související podle všeho s působením mnohočetných genů – modifikátorů, jejichž alelové frekvence se mohou u osob z různých etnických skupin podstatně lišit [90]. Riziko vzniku PN u nosičů „příznivých“ a „nepříznivých“ kombinací alelových variant celého souboru genů se může lišit trojnásobně [113]. Všechny tyto výsledky potvrzují na nové úrovni důležitou úlohu genetické složky v etiologii sporadické PN.
Mezi genetickými faktory a faktory prostředí, které určují predispozici k PN, existuje zřetelná interakce. Řadou prací byly získány údaje, jež demonstrují, že při dlouhodobém kontaktu s pesticidy je pravděpodobnost vzniku PN zvlášť vysoká u osob, které jsou nositeli nepříznivých alel genů buněčné detoxikace [18]. Byla například zjištěna vyšší patogenetická role pesticidů u špatných metabolizátorů organických sloučenin fosforu – tj. u osob, které mají nepříznivou alelu genu cytochromoxidázy CYP2D6 [119]. Analogickým způsobem se zvyšuje citlivost k pesticidům při výskytu nepříznivé varianty v promotoru genu SNCA, přičemž tento vliv je vzájemného rázu, protože riziko PN související s SNCA se modifikuje v závislosti na kontaktu s pesticidy v průběhu života [14]. Známý protektivní efekt kouření ve vztahu k riziku PN rovněž zprostředkovávají genetické faktory, například nositelství určitých alel genů GSTP1 a NAT2 [120].
Podobný obraz charakterizuje i zjištěnou asociaci PN s kraniocerebrálním traumatem. Ukázalo se, že tato asociace je nejsilnější v těch případech onemocnění, které se vyznačují pozitivní rodinnou anamnézou a/ nebo přídatným působením pesticidů a dalších známých parkinsonských neurotoxinů [81]. Poznatky z posledních let svědčí o tom, že kraniocerebrální trauma zvyšuje riziko PN pouze u osob, které jsou nositeli specifických polymorfizmů v predispozičních genech, zvláště dlouhé promotorové alely Rep1 v genu SNCA [80,121].
Ze všeho výše uvedeného vyplývá důležitý závěr: Působení environmentálních neurotoxinů, kraniocerebrálních traumat a dalších exogenních faktorů může sloužit jako spouštěč molekulárních dějů vedoucích ke vzniku PN pouze u konkrétních jedinců majících vysokou predispozici k tomuto onemocnění kvůli svému nepříznivému genetickému profilu. To dobře ilustruje interakci genetiky a prostředí a přináší důležitý klíč k porozumění mechanizmům vzniku sporadické formy PN.
Závěr
Příčiny, které vedou k poruchám zpracování α‑ synukleinu v mozkových neuronech u pacientů s PN, jsou tedy velice různorodé. Mohou souviset jak s vnějšími, tak s vnitřními faktory, jejichž těsné prolínání má rozhodující význam při vytváření rizika vzniku PN. Úloha genetiky je zvlášť velká v případech rané manifestace onemocnění, kdežto u starších osob má větší význam kombinace vlivů životního prostředí a věkově vázané nedostatečností neuronálních ochranných systémů.
Zkoumání patologie α‑ synukleinu a objevy velkého počtu genů PN umožnily stanovit klíčové molekulární mechanizmy dopaminergní neurodegenerace. V patogenezi neurodegenerativního procesu u PN mají hlavní význam dva mechanizmy:
- proteolytický stres – porucha biogeneze a proteolytické degradace neuronálních bílkovin (porucha lysozomální autofagie, mitofagie a ubikvitin‑proteazomového systému);
- porucha mitochondriální homeostázy [10,15,58,122,123].
Toto vše je doprovázeno oxidačním stresem a potlačením energetického metabolizmu neuronů. V nedávné rozsáhlé multicentrické analýze, která spojila údaje z metaanalýzy celogenomových studií genové exprese s výsledky zkoumání transkriptomu neuronů substantia nigra, bylo zjištěno spojení PN s deseti molekulárními mechanizmy bioenergetického metabolizmu, včetně metabolizmu glukózy a mitochondriální oxidační fosforylace [124]. Důležitý přídatný význam se přikládá poruchám procesů fosforylace, endozomálních funkcí a vezikulárního transportu, a rovněž aktivaci mikroglie [88].
Je třeba mít na zřeteli, že uvedené patogenetické prvky spolu vzájemně úzce souvisejí a každý z genů asociovaných s PN může přímo či nepřímo ovlivňovat četné molekulární mechanizmy. Právě tímto komplexním rázem molekulárních interakcí se vysvětlují těžkosti při vývoji metod neuroprotekce u PN. Samostatné použití kteréhokoli léčivého preparátu a/ nebo působení na jakýkoli individuální prvek patogeneze nemoci jsou málo perspektivní. Další úspěchy při odhalování etiologie PN skýtají naději na objevy principiálně nových strategií léčby tohoto těžkého neurodegenerativního onemocnění.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Přijato k recenzi: 16. 12. 2014
Přijato do tisku: 13. 4. 2015
prof. MUDr. Sergey N. Illarioškin, PhD.
Výzkumné centrum neurologie
Ruská akademie lékařských věd
Volokolamskoje šosse 80
1253 67 Moskva
Rusko
e-mail: snillario@gmail.com
Sources
1. Mizuno Y, Hattori N, Kubo S, Sato S, Nishioka K, Hatano T et al. Progress in the pathogenesis and genetics of Parkinson’s disease. Philos Trans R Soc B Biol Sci 2008; 363(1500): 2215– 2227. doi: 10.1098/ rstb.2008.2273.
2. Schiesling C, Kieper N, Seidel K, Kruger R. Review: Familial Parkinson’s disease – genetics, clinical phenotype and neuropathology in relation to the common sporadic form of the disease. Neuropathol Appl Neurobiol 2008; 34(3): 255– 271. doi: 10.1111/ j.1365‑ 2990.2008.00952.x.
3. Singleton AB, Farrer MJ, Bonifati V. The genetics of Parkinson’s disease: progress and therapeutic implications. Mov Disord 2013; 28(1): 14– 23. doi: 10.1002/ mds.25249.
4. Bartels T, Choi JG, Selkoe DJ. α‑ synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 2011; 477(7362): 107– 110. doi: 10.1038/ nature10324.
5. Abeliovich A, Schmitz Y, Fariñas I, Choi‑ Lundberg D, Ho WH, Castillo PE et al. Mice lacking alphα‑ synuclein display functional deficits in the nigrostriatal system. Neuron 2000; 25(1): 239– 252.
6. Lee FJ, Liu F, Pristupa ZB, Niznik HB. Direct binding and functional coupling of alphα‑ synuclein to the dopamine transporters accelerate dopamine‑induced apoptosis. Faseb J 2001; 15(6): 916– 926.
7. Murphy DD, Rueter SM, Trojanowski JQ, Lee VM. Synucleins are developmentally expressed, and alphα‑ synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J Neurosci 2000; 20(9): 3214– 3220.
8. Breydo L, Wu JW, Uversky VN. α‑ synuclein misfolding and Parkinson‘s disease. Biochim Biophys Acta 2012; 1822(2): 261– 285. doi: 10.1016/ j.bbadis.2011.10.002.
9. Jenner P, Morris HR, Robbins TW, Goedert M, Hardy J,Ben‑ Shlomo Y et al. Parkinson’s disease – the debate on the clinical phenomenology, etiology, pathology and pathogenesis. J Parkinsons Dis 2013; 3(1): 1– 11. doi: 10.3233/ JPD‑ 130175.
10. Lim KL, Zhang CW. Molecular events underlying Parkinson‘s disease – an interwoven tapestry. Frontiers Neurol 2013; 4: 33. doi: 10.3389/ fneur.2013.00033.
11. Obeso JA, Rodriguez‑ Oroz MC, Goetz CG, Marin C, Kordower JH, Rodriguez M et al. Missing pieces in the Parkinson’s disease puzzle. Nat Med 2010; 16(6): 653– 661. doi: 10.1038/ nm.2165.
12. Saiki S, Sato S, Hattori N. Molecular pathogenesis of Parkinson‘s disease: update. J Neurol Neurosurg Psychiatry 2012; 83(4): 430– 436. doi: 10.1136/ jnnp‑ 2011‑ 301205.
13. Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. Alphα‑ synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci USA 1998; 95(11): 6469– 6473.
14. Gatto NM, Rhodes SL, Manthripragada AD, Bronstein J,Cockburn M, Farrer M et al. α‑ synuclein gene may interact with environmental factors in increasing risk of Parkinson’s disease. Neuroepidemiology 2010; 35(3): 191– 195. doi: 10.1159/ 000315157.
15. Lin MK, Farrer MJ. Genetics and genomics of Parkinson‘s disease. Genome Med 2014; 6(6): 48. doi: 10.1186/ gm566.
16. Schapira AH, Jenner P. Etiology and pathogenesis of Parkinson‘s disease. Mov Disord 2011; 26(6): 1049– 1055. doi: 10.1002/ mds.23732.
17. Vargas KJ, Makani S, Davis T, Westphal CH, Castillo PE, Chandra SS. Synucleins regulate the kinetics of synaptic vesicle endocytosis. J Neurosci 2014; 34(28): 9364– 9376. doi: 10.1523/ JNEUROSCI.4787‑ 13.2014.
18. Veldman BA, Wijn AM, Knoers N, Praamstra P, Horstink MW. Genetic and environmental risk factors in Parkinson’s disease. Clin Neurol Neurosurg 1998; 100(1): 15– 26.
19. Menšíková K, Kaňovský P, Kaiserová M, Nestrašil I, Bareš M. Proměnlivá tvář parkinsonské neurodegenerace. Cesk Slov Neurol N 2013; 76/ 109(1): 26– 34.
20. Priyadarshi A, Khuder SA, Schaub EA, Priyadarshi SS.Environmental risk factors and Parkinson’s disease: a meta‑analysis. Environ Res 2001; 86(2): 122– 127.
21. Wirdefeldt K, Adami HO, Cole P, Trichopoulos D, Mandel J. Epidemiology and etiology of Parkinson‘s disease: a review of the evidence. Eur J Epidemiol 2011; 26 (Suppl 1):S1– S58. doi: 10.1007/ s10654‑ 011‑ 9581‑ 6.
22. Betarbet R, Sherer TB, MacKenzie G, Garcia‑ Osuna M, Panov AV, Greenamyre JT et al. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci 2000; 3(12): 1301– 1306.
23. Meredith GE, Sonsalla PK, Chesselet MF. Animal models of Parkinson’s disease progression. Acta Neuropathol 2008; 115(4): 385– 398. doi: 10.1007/ s00401‑ 008‑ 0350‑ x.
24. Ossowska K, Smiałowska M, Kuter K, Wierońska J, Zieba B, Wardas J et al. Degeneration of dopaminergic mesocortical neurons and activation of compensatory processes induced by a long‑term paraquat administration in rats: implications for Parkinson‘s disease. Neuroscience 2006; 141(4): 2155– 2165.
25. Dick FD, De Palma G, Ahmadi A, Scott NW, Prescott GJ,Bennett J et al. Environmental risk factors for Parkinson’s disease and parkinsonism: the Geoparkinson study. Occup Environ Med 2007; 64(10): 666– 672.
26. Gatto NM, Cockburn M, Bronstein J, Manthripragada AD,Ritz B. Well‑water consumption and Parkinson’s disease in rural California. Environ Health Persp 2009; 117(12): 1912– 1918. doi: 10.1289/ ehp.0900852.
27. Gorell JM, Johnson CC, Rybicki BA, Peterson EL, Richardson RJ. The risk of Parkinson’s disease with exposure to pesticides, farming, well water, and rural living. Neurology 1998; 50(5): 1346– 1350.
28. Kamel F, Tanner C, Umbach D, Hoppin J, Alavanja M, Blair A et al. Pesticide exposure and self‑ reported Parkinson’s disease in the agricultural health study. Am J Epidemiol 2007; 165(4): 364– 374.
29. Costello S, Cockburn M, Bronstein J, Zhang X, Ritz B. Parkinson‘s disease and residential exposure to maneb and paraquat from agricultural applications in the central valley of California. Am J Epidemiol 2009; 169(8): 919– 926. doi: 10.1093/ aje/ kwp006.
30. Tanner CM, Kamel F, Ross GW, Hoppin JA, Goldman SM,Korell M et al. Rotenone, paraquat, and Parkinson‘s disease. Environ Health Perspect 2011; 119(6): 866– 872. doi: 10.1289/ ehp.1002839.
31. Corrigan FM, Wienburg CL, Shore RF, Daniel SE, Mann D.Organochlorine insecticides in substantia nigra in Parkinson‘s disease. J Toxicol Environ Health A 2000; 59(4): 229– 234.
32. Fleming L, Mann JB, Bean J, Briggle T, Sanchez‑ Ramos JR.Parkinson‘s disease and brain levels of organochlorine pesticides. Ann Neurol 1994; 36(1): 100– 103.
33. Chorfa A, Lazizzera C, Bétemps D, Morignat E, Dussurgey S, Andrieu T et al. A variety of pesticides trigger in vitro α‑ synuclein accumulation, a key event in Parkinson‘s disease. Arch Toxicol 2014 [Epub ahead of print].
34. Uversky VN, Li J, Fink AL. Pesticides directly accelerate the rate of alphα‑ synuclein fibril formation: a possible factor in Parkinson‘s disease. FEBS Lett 2001; 500(3): 105– 108.
35. Franco R, Li SM, Rodriguez‑ Rocha H, Burns M, Panayiotidis MI. Molecular mechanisms of pesticide‑induced neurotoxicity: relevance to Parkinson‘s disease. Chem Biol Interact 2010; 188(2): 289– 300. doi: 10.1016/ j.cbi.2010.06.003.
36. Langston JW, Forno LS, Tetrud J, Reeves AG, Kaplan JA, Karluk D. Evidence of active nerve cell degeneration in the substantia nigra in humans years after 1- methyl4- phenyl‑ 1,2,3,6- tetrahydropyridine exposure. Ann Neurol 1999; 46(4): 598– 605.
37. Perese DA, Ulman J, Viola J, Ewing SE, Bankiewicz KS. A 6- hydroxydopamine‑induced selective parkinsonian rat model. Brain Res 1989; 494(2): 285– 293.
38. Powers KM, Smith‑ Weller T, Franklin GM, Longstreth WT jr, Swanson PD, Checkoway H. Parkinson’s disease risks associated with dietary iron, manganese, and other nutrient intakes. Neurology 2003; 60(11): 1761– 1766.
39. Uversky VN, Li J, Bower K, Fink AL. Synergistic effects of pesticides and metals on the fibrillation of alphα‑ synuclein: implications for Parkinson‘s disease. Neurotoxicology 2002; 23(4– 5): 527– 536.
40. Werneck AL, Alvarenga H. Genetics, drugs and environmental factors in Parkinson’s disease. A case‑ control study. Arq Neuropsiquiatr 1999; 57(2B): 347– 355.
41. Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN,Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 2003; 24(2): 197– 211.
42. Del Tredici K, Braak H. Lewy pathology and neurodegeneration in premotor Parkinson‘s disease. Mov Disord 2012; 27(5): 597–607. doi: 10.1002/ mds.24921.
43. Adler CH, Dugger BN, Hinni ML, Lott DG, Driver‑ Dunckley E, Hidalgo J et al. Submandibular gland needle biopsy for the diagnosis of Parkinson disease. Neurology 2014; 82(10): 858– 864. doi: 10.1212/ WNL.0000000000000204.
44. Beach TG, Adler CH, Sue LI, Vedders L, Lue L, White CL et al. Multi‑organ distribution of phosphorylated α‑ synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol 2010; 119(6): 689– 702. doi: 10.1007/ s00401‑ 010‑ 0664‑ 3.
45. Folgoas E, Lebouvier T, Leclair‑ Visonneau L, Cersosimo MG, Barthelaix A, Derkinderen P et al. Diagnostic value of minor salivary glands biopsy for the detection of Lewy pathology. Neurosci Lett 2013; 551: 62– 64. doi: 10.1016/ j.neulet.2013.07.016.
46. Lebouvier T, Neunlist M, Bruley des Varannes S, Coron E,Drouard A, N‘Guyen JM, Chaumette T et al. Colonic biopsies to assess the neuropathology of Parkinson’s disease and its relationship with symptoms. PLoS One 2010; 5(9): e12728. doi: 10.1371/ journal.pone.0012728.
47. Phillips RJ, Walter GC, Wilder SL, Baronowsky EA, Powley TL. Alphα‑ synuclein‑immunopositive myenteric neurons and vagal preganglionic terminals: autonomic pathway implicated in Parkinson’s disease? Neuroscience 2008; 153(3): 733– 750. doi: 10.1016/ j.neuroscience.2008.02.074.
48. Paillusson S, Clairembault T, Biraud M, Neunlist M, Derkinderen P. Activity‑ dependent secretion of alhα‑ synuclein by enteric neurons. J Neurochem 2013; 125(4): 512– 517. doi: 10.1111/ jnc.12131.
49. Jellinger KA. Synuclein deposition and non‑motor symptoms in Parkinson disease. J Neurol Sci 2011; 310(1– 2): 107– 111. doi: 10.1016/ j.jns.2011.04.012.
50. Hellenbrand W, Seidler A, Boeing H, Robra BP, Vieregge P, Nischan P et al. Diet and Parkinson’s disease. I: a possible role for the past intake of specific foods and food groups. Results from a self‑ administered food‑ frequency questionnaire in a case‑ control study. Neurology 1996; 47(3): 636– 643.
51. Ross GW, Petrovitch H. Current evidence for neuroprotective effects of nicotine and caffeine against Parkinson’s disease. Drugs Aging 2001; 18(11): 797– 806.
52. Tan LC, Koh WP, Yuan JM, Wang R, Au WL, Tan JH et al. Differential effects of black versus green tea on risk of Parkinson’s disease in the Singapore Chinese Health Study. Am J Epidemiol 2008; 167(5): 553– 560.
53. Hong DP, Fink AL, Uversky VN. Smoking and Parkinson’s disease: does nicotine affect alphα‑ synuclein fibrillation? Biochim Biophys Acta 2009; 1794(2): 282– 290. doi: 10.1016/ j.bbapap.2008.09.026.
54. Olanow CW. Do prions cause Parkinson disease? The evidence accumulates. Ann Neurol 2014; 75(3): 331– 333. doi: 10.1002/ ana.24098.
55. Olanow CW, McNaught K. Parkinson’s disease, proteins, and prions: milestones. Mov Disord 2011; 26(6): 1056– 1071. doi: 10.1002/ mds.23767.
56. Luk KC, Song C, O‘Brien P, Stieber A, Branch JR, Brunden KR et al. Exogenous alpha‑ synuclein fibrils seed the formation of Lewy body‑like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A 2009; 106(47): 20051– 20056. doi: 10.1073/ pnas.0908005106.
57. Volpicelli‑ Daley L, Luk K, Patel T, Tanik S, Riddle D, Stieber A et al. Exogenous α‑ synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 2011; 72(1): 57– 71. doi: 10.1016/ j.neuron.2011.08.033.
58. Moore DJ, West AB, Dawson VL, Dawson TM. Molecular pathophysiology of Parkinson’s disease. Ann Rev Neurosci 2005; 28: 57– 87.
59. Conway KA, Harper JD, Lansbury PT jr. Fibrils formed in vitro from α‑ synuclein and two mutant forms linked to Parkinson’s disease are typical amyloid. Biochemistry 2000; 39(10): 2552– 2563.
60. Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body‑like pathology in long‑ term embryonic nigral transplants in Parkinson‘s disease. Nat Med 2008; 14(5): 504– 506. doi: 10.1038/ nm1747.
61. Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJet al. Lewy bodies in grafted neurons in subjects with Parkinson‘s disease suggest host‑to‑ graft disease propagation. Nat Med 2008; 14(5): 501– 503. doi: 10.1038/ nm1746.
62. Danzer KM, Kranich LR, Ruf WP, Cagsal‑ Getkin O, Winslow AR, Zhu L et al. Exosomal cell‑ to‑ cell transmission of α‑ synuclein oligomers. Mol Neurodegener 2012; 7: 42. doi: 10.1186/ 1750‑ 1326‑ 7‑ 42.
63. Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L et al. Inclusion formation and neuronal cell death through neuron‑ to‑ neuron transmission of alphα‑ synuclein. Proc Natl Acad Sci USA 2009; 106(31): 13010– 13015. doi: 10.1073/ pnas.0903691106.
64. Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH et al. Cell‑ produced α‑ synuclein is secreted in a calcium‑ dependent manner by exosomes and impacts neuronal survival. J Neurosci 2010; 30(20): 6838– 6851. doi: 10.1523/ JNEUROSCI.5699‑ 09.2010.
65. Angot E, Steiner JA, Hansen C, Li JY, Brundin P. Are synucleinopathies prion‑like disorders? Lancet Neurol 2010; 9(11): 1128– 1138. doi: 10.1016/ S1474‑ 4422(10)70213‑ 1.
66. Tokuda T, Qureshi MM, Ardah MT, Varghese S, Shehab SA, Kasai T et al. Detection of elevated levels of alphα‑ synuclein oligomers in CSF from patients with Parkinson disease. Neurology 2010; 75(20): 1766– 1772. doi: 10.1212/ WNL.0b013e3181fd613b.
67. Luk KC, Lee VM. Modeling Lewy pathology propagation in Parkinson‘s disease. Parkinsonism Relat Disord 2014; 20 (Suppl 1): S85– S87. doi: 10.1016/ S1353‑ 8020(13)70022‑ 1.
68. Luk K, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski J et al. Pathological α‑ synuclein transmission initiates Parkinson‑like neurodegeneration in non‑transgenic mice. Science 2012; 338(6109): 949– 953. doi: 10.1126/ science.1227157.
69. Masuda‑ Suzukake M, Nonaka T, Hosokawa M, Oikawa T,Arai T, Akiyama H et al. Prion‑like spreading of pathological α‑ synuclein in brain. Brain 2013; 136(4): 1128– 1138. doi: 10.1093/ brain/ awt037.
70. Luk KC, Kehm VM, Zhang B, O‘Brien P, Trojanowski JQ, Lee VM. Intracerebral inoculation of pathological α‑ synuclein initiates a rapidly progressive neurodegenerative α‑ synucleinopathy in mice. J Exp Med 2012; 209(5): 975– 986. doi: 10.1084/ jem.20112457.
71. Mougenot AL, Nicot S, Bencsik A, Morignat E, Verchère J, Lakhdar L et al. Prion‑like acceleration of a synucleinopathy in a transgenic mouse model. Neurobiol Aging 2012; 33(9): 2225– 2228. doi: 10.1016/ j.neurobiolaging.2011.06.022.
72. Recasens A, Dehay B, Bové J, Carballo‑ Carbajal I, Dovero S, Pérez‑ Villalba A et al. Lewy body extracts from Parkinson’s disease brains trigger α‑ synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol 2014; 75(3): 351– 362. doi: 10.1002/ ana.24066.
73. Watts JC, Giles K, Oehler A, Middleton L, Dexter DT, Gentleman SM et al. Transmission of multiple system atrophy prions to transgenic mice. Proc Natl Acad Sci USA 2013; 110(48): 19555–19560.
74. Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang Bet al. Distinct alphα‑ synuclein strains differentially promote tau inclusions in neurons. Cell 2013; 154(1): 103– 117. doi: 10.1016/ j.cell.2013.05.057.
75. Guo JL, Lee VMY. Cell‑ to‑ cell transmission of pathogenic proteins in neurodegenerative diseases. Nat Med 2014; 20(2): 130– 138. doi: 10.1038/ nm.3457.
76. Jucker M, Walker LC. Self‑ propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013; 501(7465): 45– 51. doi: 10.1038/ nature12481.
77. Irwin DJ, Abrams JY, Schonberger LB, Leschek EW, Mills JL, Lee VM et al. Evaluation of potential infectivity of Alzheimer and Parkinson disease proteins in recipients of cadaver‑ derived human growth hormone. JAMA Neurol 2013; 70(4): 462– 468. doi: 10.1001/ jamaneurol.2013.1933.
78. Tran HT, Chung CH, Iba M, Zhang B, Trojanowski JQ, Luk KC et al. α‑ synuclein immunotherapy blocks uptake and templated propagation of misfolded α‑ synuclein and neurodegeneration. Cell Rep 2014; 7(6): 2054– 2065. doi: 10.1016/ j.celrep.2014.05.033.
79. Jordan BD. Chronic traumatic brain injury associated with boxing. Semin Neurol 2000; 20(2): 179– 185.
80. Gershanik OS. Trauma and Parkinson’s disease. Handb Clin Neurol 2007; 84: 487– 499. doi: 10.1016/ S0072‑ 9752(07)84057‑ 7.
81. Taylor CA, Saint‑ Hilaire MH, Cupples LA, Thomas CA, Burchard AE, Feldman RG et al. Environmental, medical, and family history risk factors for Parkinson’s disease: a New England‑based case control study. Am J Med Genet 1999; 88(6): 742– 749.
82. Jafari S, Etminan M, Aminzadeh F, Samii A. Head injury and risk of Parkinson disease: a systematic review and meta‑analysis. Mov Disord 2013; 28(9): 1222– 1229. doi: 10.1002/ mds.25458.
83. Irwin DJ, Trojanowski JQ. Many roads to Parkinson’s disease neurodegeneration: head trauma – a road more traveled than we know? Mov Disord 2013; 28(9): 1167– 1170. doi: 10.1002/ mds.25551.
84. Uryu K, Giasson BI, Longhi L, Martinez D, Murray I, Conte V et al. Age‑ dependent synuclein pathology following traumatic brain injury in mice. Exp Neurol 2003; 184(1): 214– 224.
85. Kasten M, Chade A, Tanner CM. Epidemiology of Parkinson’s disease. Handb Clin Neurol 2007; 83: 129– 151. doi: 10.1016/ S0072‑ 9752(07)83006‑ 5.
86. Schulz JB, Lindenau J, Seyfried J, Dichgans J. Glutathione, oxidative stress and neurodegeneration. Eur J Biochem 2000; 267(16): 4904– 4911.
87. Sulzer D, Surmeier DJ. Neuronal vulnerability, pathogenesis, and Parkinson‘s disease. Mov Disord 2013; 28(6): 41– 50. doi: 10.1002/ mds.25095.
88. Chai C, Lim KL. Genetic insights into sporadic Parkinson’s disease pathogenesis. Curr Genomics 2013; 14(8): 486– 501. doi: 10.2174/ 1389202914666131210195808.
89. Piccini P, Burn DJ, Ceravolo R, Maraganore D, Brooks DJ.The role of inheritance in sporadic Parkinsons’ disease: evidence from a longitudinal study of dopaminergic function in twins. Ann Neurol 1999; 45(5): 577– 582.
90. Simón‑ Sánchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D et al. Genome‑ wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet 2009; 41(12): 1308– 1312. doi: 10.1038/ ng.487.
91. Bonifati V. Genetics of Parkinson‘s disease – state of the art, 2013. Parkinsonism Relat Disord 2014; 20 (Suppl 1): S23– S28. doi: 10.1016/ S1353‑ 8020(13)70009‑ 9.
92. Ibáñez P, Lesage S, Janin S, Lohmann E, Durif F, Destée A et al. Alphα‑ synuclein gene rearrangements in dominantly inherited parkinsonism: frequency, phenotype, and mechanisms. Arch Neurol 2009; 66(1): 102– 108. doi: 10.1001/ archneurol.2008.555.
93. Kilarski LL, Pearson JP, Newsway V, Majounie E, Knipe MD, Misbahuddin A et al. Systematic review and UK‑based study of PARK2 (parkin), PINK1, PARK7 (DJ‑ 1) and LRRK2 in early‑ onset Parkinson’s disease. Mov Disord 2012; 27(12): 1522– 1529. doi: 10.1002/ mds.25132.
94. Wang Y, Clark LN, Louis ED, Mejia‑ Santana H, Harris J, Cote LJ et al. Risk of Parkinson disease in carriers of parkin mutations:estimation using the kin‑cohort method. Arch Neurol 2008; 65(4): 467– 474. doi: 10.1001/ archneur.65.4.467.
95. Lim KL, Ng XH, Grace LG, Yao TP. Mitochondrial dynamics and Parkinson’s disease: focus on parkin. Antioxid Redox Signal 2012; 16(9): 935– 949. doi: 10.1089/ ars.2011.4105.
96. Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 2008; 183(5): 795– 803. doi: 10.1083/ jcb.200809125.
97. Canet‑ Avilés RM, Wilson MA, Miller DW, Ahmad R, McLendon C, Bandyopadhyay S et al. The Parkinson’s disease protein DJ‑ 1 is neuroprotective due to cyctein‑sulfinic acid‑driven mitochondrial localization. Proc Natl Acad Sci U S A 2004; 101(24): 9103– 9108.
98. Thomas KJ, McCoy MK, Blackinton J, Beilina A, van der Brug M, Sandebring A et al. DJ‑ 1 acts in parallel to the PINK1/ parkin pathway to control mitochondrial function and autophagy. Hum Mol Genet 2011; 20(1): 40– 50. doi: 10.1093/ hmg/ ddq430.
99. Healy DG, Falchi M, O‘Sullivan SS, Bonifati V, Durr A, Bressman S et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2‑associated Parkinson’s disease: a case‑ control study. Lancet Neurol 2008; 7(7): 583– 590. doi: 10.1016/ S1474‑ 4422(08)70117‑ 0.
100. Kračunová K, Kovačovicová M, Baldovič M, Valkovič P, Kádaši L, Benetin J. The Incidence of Mutation on the Leucine‑ rich Repeat Kinase 2 Gene in Patients with Parkinson’s Disease in Slovakia. Cesk Slov Neurol N 2011; 74/ 107(4): 443– 445.
101. Vilariño‑ Güell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ et al. VPS35 mutations in Parkinson disease. Am J Hum Genet 2011; 89(1): 162– 167. doi: 10.1016/ j.ajhg.2011.06.001.
102. Zimprich A, Benet‑ Pagès A, Struhal W, Graf E, Eck SH, Offman MN et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late‑ onset Parkinson disease. Am J Hum Genet 2011; 89(1): 168– 175. doi: 10.1016/ j.ajhg.2011.06.008.
103. Sharma M, Ioannidis JP, Aasly JO, Annesi G, Brice A,Bertram L et al. A multi‑centre clinico‑ genetic analysis of the VPS35 gene in Parkinson disease indicates reduced penetrance for disease‑associated variants. J Med Genet 2012; 49(11): 721– 726. doi: 10.1136/ jmedgenet‑ 2012‑ 101155.
104. Kay DM, Factor SA, Samii A, Higgins DS, Griffith A, Roberts JW et al. Genetic association between alphα‑ synuclein and idiopathic Parkinson’s disease. Am J Med Genet B Neuropsychiatr Genet 2008; 147B(7): 1222– 1230. doi: 10.1002/ ajmg.b.30758.
105. Maraganore DM, de Andrade M, Elbaz A, Farrer MJ,Ioannidis JP, Krüger R et al. Collaborative analysis of alphα‑ synuclein gene promoter variability and Parkinson disease. JAMA 2006; 296(6): 661– 670.
106. Parsian AJ, Racette BA, Zhao JH, Sinha R, Patra B, Perlmutter JS et al. Association of alphα‑ synuclein gene haplotypes with Parkinson’s disease. Parkinsonism Relat Disord 2007; 13(6): 343– 347.
107. Mueller JC, Fuchs J, Hofer A, Zimprich A, Lichtner P, Illig T et al. Multiple regions of alphα‑ synuclein are associated with Parkinson’s disease. Ann Neurol 2005; 57(4): 535– 541.
108. Rhinn H, Qiang L, Yamashita T, Rhee D, Zolin A, Vanti Wet al. Alternative alphα‑ synuclein transcript usage as a convergent mechanism in Parkinson’s disease pathology. Nat Commun 2012; 3: 1084. doi: 10.1038/ ncomms2032.
109. Sidransky E, Nalls MA, Aasly JO, Aharon‑ Peretz J, Annesi G, Barbosa ER et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med 2009; 361(17): 1651– 1661. doi: 10.1056/ NEJMoa0901281.
110. Westbroek W, Gustafson AM, Sidransky E. Exploring the link between glucocerebrosidase mutations and parkinsonism. Trends Mol Med 2011; 17(9): 485– 493. doi: 10.1016/ j.molmed.2011.05.003.
111. Alonso‑ Navarro H, Jimenez‑ Jimenez FJ, Garcia‑ Martin E, Agundez JA. Genomic and pharmacogenomics biomarkers of Parkinson’s disease. Curr Drug Metab 2014; 15(2): 129– 181.
112. International Parkinson Disease Genomics Consortium. Imputation of sequence variants for identification of genetic risks for Parkinson‘s disease: a meta‑analysis of genome‑ wide association studies. Lancet 2011; 377(9766): 641– 649. doi: 10.1016/ S0140‑ 6736(10)62345‑ 8.
113. International Parkinson‘s Disease Genomics Consortium (IPDGC); Wellcome Trust Case Control Consortium 2 (WTCCC2). A two‑stage meta‑analysis identifies several new loci for Parkinson‘s disease. PLoS Genet 2011; 7(6): e1002142. doi: 10.1371/ journal.pgen.1002142.
114. Lill CM, Roehr JT, McQueen MB, Kavvoura FK, Bagade S, Schjeide BM et al. Comprehensive research synopsis and systematic meta‑analyses in Parkinson’s disease genetics: the PDGene database. PLoS Genet 2012; 8(3): e1002548. doi: 10.1371/ journal.pgen.1002548.
115. Pastor P. Genetic heterogeneity in Parkinson disease: the meaning of GWAS and replication studies. Neurology 2012; 79(7): 619– 620. doi: 10.1212/ WNL.0b013e318264e3d2.
116. Imamura K, Hishikawa N, Sawada M, Nagatsu T, Yoshida M, Hashizume Y. Distribution of major histocompatibility complex class II‑ positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol 2003; 106(6): 518– 526.
117. Ouchi Y, Yoshikawa E, Sekine Y, Futatsubashi M, Kanno T, Ogusu T et al. Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann Neurol 2005; 57(2): 168– 175.
118. Moehle MS, Webber PJ, Tse T, Sukar N, Standaert DG, DeSilva TM et al. LRRK2 inhibition attenuates microglial inflammatory responses. J Neurosci 2012; 32(5): 1602– 1611. doi: 10.1523/ JNEUROSCI.5601‑ 11.2012.
119. Elbaz A, Levecque C, Clavel J, Vidal JS, Richard F, Amouyel P et al. CYP2D6 polymorphism, pesticide exposure, and Parkinson’s disease. Ann Neurol 2004; 55(3): 430– 434.
120. De Palma G, Dick FD, Calzetti S, Scott NW, Prescott GJ,Osborne A et al. A case‑ control study of Parkinson‘s disease and tobacco use: gene‑ tobacco interactions. Mov Disord 2010; 25(7): 912– 919. doi: 10.1002/ mds.22980.
121. Goldman SM, Kamel F, Bhudikanok F. Alphα‑ synuclein (SNCA) genotype modifies the association between head injury and Parkinson’s disease (PD). Neurology 2009; 72 (Suppl 3): S23.005.
122. Hirsch EC, Jenner P, Przedborski S. Pathogenesis of Parkinson’s disease. Mov Disord 2013; 28(1): 24– 30. doi: 10.1002/ mds.25032.
123. Rubinsztein DC. The roles of intracellular protein‑degradation pathways in neurodegeneration. Nature 2006; 443(7113): 780– 786.
124. Zheng B, Liao Z, Locascio JJ, Lesniak KA, Roderick SS, Watt ML et al. PGC‑ 1α, a potential therapeutic target for early intervention in Parkinson‘s disease. Sci Transl Med 2010; 2(52): 52– 73. doi: 10.1126/scitranslmed.3001059.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2015 Issue 3
Most read in this issue
- Addenbrookský kognitivní test – orientační normy pro českou populaci
- Míšní šok – od patofyziologie ke klinickým projevům
- Diagnostika epileptických záchvatů
- Vzduchová embolie mozku – kazuistika