
Absces mozku jako první klinická manifestace hereditární hemoragické teleangiektázie – tři kazuistiky
Brain Abscess as the First Clinical Manifestation of Hereditary Hemorrhagic Telangiectasia – Three Case Reports
Hereditary hemorrhagic telangiectasia or Rendu‑ Osler‑ Weber disease is an autosomal‑ dominant disorder that results in fibrovascular dysplasia and is characterized by telangiectases and arteriovenous malformation of the skin, mucosa and viscera. Pulmonary arteriovenous malformations as a source of septic emboli may give rise to brain abscess that so often is the first and only clinical manifestation of hereditary hemorrhagic telangiectasia. Three cases of patients suffering from this disease with brain abscess as the first and serious symptom are presented. Since this is a relatively rare disease, it is not always considered in the differential diagnosis and thus not diagnosed. This disease has to be considered in patients with brain abscess and, in case of a suspicion, it must be properly examined. When pulmonary arteriovenous malformation is verified, it has to be treated in order to prevent recurrent brain abscesses. Care of these patients should be comprehensive and multidisciplinary.
Key words:
hereditary hemorrhagic telangiectasia – Rendu-Osler-Weber disease – brain abscess – arteriovenous malformation – epistaxis – endoglin
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Authors:
G. Hanoun 1; V. Vybíhal 1
![]() ; Marek Sova 1
; Marek Sova 1
![]() ; J. Boudný 2; T. Svoboda 1; M. Smrčka 1
; J. Boudný 2; T. Svoboda 1; M. Smrčka 1
Authors‘ workplace:
LF MU a FN Brno
Neurochirurgická klinika
1; LF MU a FN Brno
Radiologická klinika
2
Published in:
Cesk Slov Neurol N 2015; 78/111(3): 348-353
Category:
Case Report
Overview
Hereditární hemoragická teleangiektázie neboli morbus Rendu‑ Osler‑ Weber je fibrovaskulární dysplazie s autozomálně dominantní dědičností charakteristická přítomností teleangiektázií a arteriovenózních malformací kůže, sliznic a vnitřních orgánů. Plicní arteriovenózní malformace jako zdroj septických embolů mohou zapříčinit vznik mozkového abscesu, který tak bývá prvním a často jediným projevem heriditární hemoragické teleangiektázie. V práci jsou prezentovány tři kazuistiky pacientů trpících touto chorobou, kde prvním a závažným příznakem byl vzniklý mozkový absces. Protože se jedná o relativně vzácné onemocnění, nebývá vždy zvažováno v rámci diferenciální diagnostiky a tím pádem ani diagnostikováno. U každého pacienta s mozkovým abscesem je nezbytné na toto onemocnění pomýšlet a v případě podezření je nutné jej řádně vyšetřit. Při verifikované plicní arteriovenózní malformaci je třeba ji ošetřit a zabránit tak vzniku recidivujících mozkových abscesů. Péče o tyto pacienty je komplexní a multioborová.
Klíčová slova:
hereditární hemoragická teleangiektázie –morbus Rendu-Osler-Weber – absces mozku – arteriovenózní malformace – epistaxe – endoglin
Úvod
Hereditární hemoragická teleangiektázie (HHT) neboli morbus Rendu‑ Osler‑ Weber je fibrovaskulární dysplazie s autozomálně dominantní dědičností charakteristická přítomností teleangiektázií a arteriovenózních malformací (AVM) kůže, sliznic a vnitřních orgánů [1].
První dosud známý popis HHT pochází od Suttona z roku 1864 [2]. V roce 1896 Henry Rendu upřesnil popis onemocnění, odlišil hemofilii a popsal spojitost mezi epistaxemi a výskytem malých kožních a slizničních teleangiektázií. V roce 1901 poukázal Sir William Osler na rodinný výskyt onemocnění a na možnost viscerální manifestace. O šest let později Frederik Parkes Weber doplnil popis onemocnění o hereditární angiomy, jejich spojitost s opakovaným krvácením a jeho lokální léčbu adrenalinem. Termín HHT se v literatuře objevuje poprvé v roce 1909, kdy jej použil Frederic Hanes.
Předpokládaná prevalence onemocnění je mezi 1 : 2 500 až 1 : 40 000 v celosvětovém měřítku s výskytem v Evropě okolo 1 : 8 000 [3– 5]. Porucha vývoje cév a přítomnost mnohočetných AVM jsou charakteristickým znakem pro HHT. Malé AVM nebo spíše teleangiektázie postihují kůži, sítnice, spojivky a sliznice. Fragilita cév a jejich povrchové uložení vede často ke krvácení. Epistaxe je popisována až u 90 % pacientů s HHT [6]. Velké AVM jsou nejčastěji přítomny v plicích, mozku, páteři, gastrointestinálním traktu a v játrech. Plicní AVM jsou možným zdrojem embolů s rizikem vzniku iktu a v případě septických embolů zdrojem abscesů mozku. Tato závažná komplikace může být vzácně i prvním projevem HHT u pacientů s asymptomatickou plicní AVM [7].
Teleangiektázie a AVM vznikají pravděpodobně v důsledku poruchy angiogeneze. Přesný mechanizmus, kterým mutace genů ovlivňuje angiogenezi, není dosud jasný. Patrně dochází k narušení rovnováhy mezi pro‑ a antiangiogenními signály v cévách. Vývoj nových krevních cév vyžaduje aktivaci a migraci různých typů buněk, hlavně endotelu hladké svaloviny a pericytů. Ztráta svalové vrstvy, narušení elasticity cévní stěny a změny v endotelu cév mohou vést k vyšší fragilitě cév a následně i ke vzniku krvácení [8].
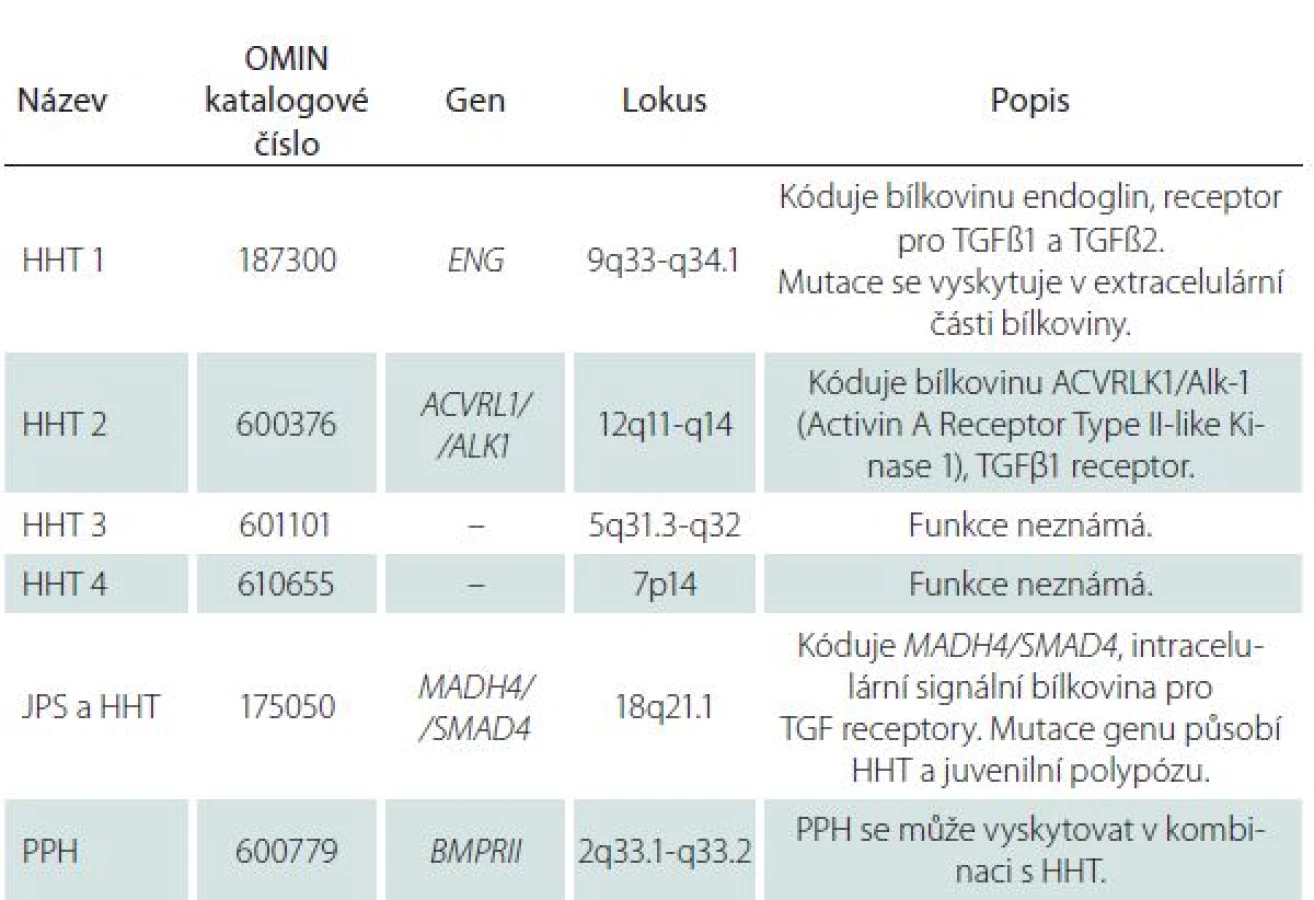
U 60– 80 % nemocných s HHT byla zjištěna mutace jednoho z minimálně dvou genů, endoglinu (ENG) nebo genu kódujícího aktivin receptoru podobnou kinázu 1 (ACVRL1), a to v poměru 53 : 47. Mutace genu ENG na chromozomu 9q33– q34.1 kódujícího stejnojmennou bílkovinu, receptor pro transformující růstový faktor beta (TGFβ1 a TGFβ3), který je součástí glykoproteinové membrány, se vyskytuje u HTT prvního typu. Mutace genu ACVRL1 na chromozomu 12q11– q14 kódujícího stejnojmennou kinázu se vyskytuje u HTT druhého typu. TGFβ má vliv na angiogenezi, na tkáňovou reparaci a reguluje funkce endoteliálních buněk stimulací nebo inhibicí jejich proliferace [8]. Další mutace se vyskytují velice zřídka (tab. 1).
V roce 2000 byla stanovena tzv. Curaçao kritéria pro klinickou diagnózu HHT (tab. 2) [1]. Diagnóza HHT je jistá přítomností alespoň třech výše uvedených kritérií a pravděpodobná přítomností dvou kritérií. Klinické projevy HHT souvisí s postiženým orgánem, s oblastí výskytu a velikostí AVM (tab. 3).


Často bývá prvním projevem onemocnění závažná komplikace, jakou je mozkový absces, pro kterou je pacient hospitalizován. Autoři článku prezentují tři kazuistiky pacientů s HHT s takovým průběhem.
Kazuistika 1
Žena (33 let) byla přijata na neurologii pro parciální levostranné motorické jacksonské paroxyzmy se sekundární generalizací. Otec je sledován pro HHT, anémii a je po operaci kyčle pro absces. Pacientka udávala recidivující epistaxe. Na výpočetní tomografii (CT) se zobrazila cystická expanze v pravém parietálním laloku. Nález byl verifikován na magnetické rezonanci (MR) s vyslovením podezření na nádor (difuzně vážené snímky nebyly tehdy k dispozici). Proto byla zvažována stereotaktická biopsie.
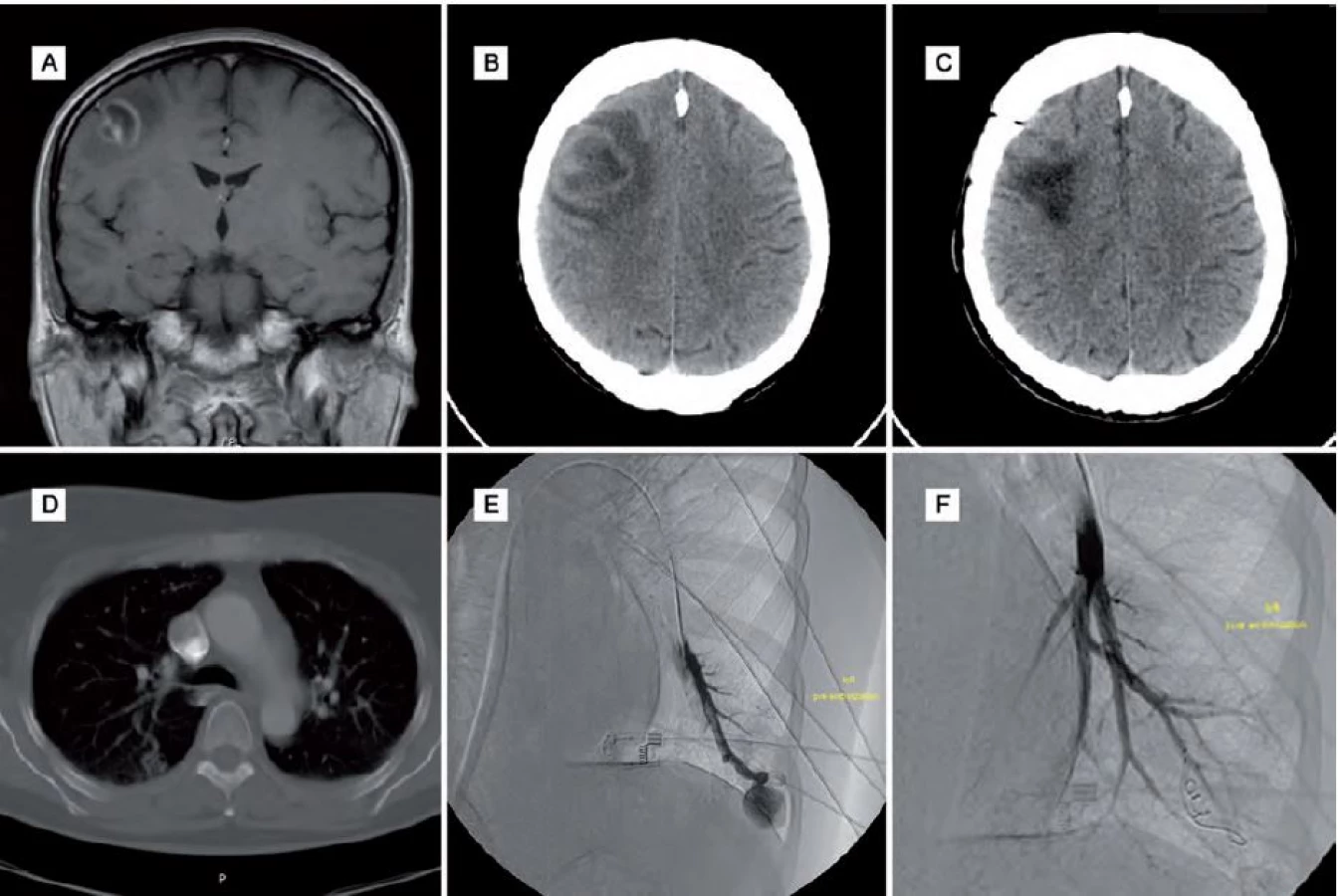
V dalším průběhu narůstala cefalea, zhoršovala se řeč a hybnost levé horní končetiny. Po týdnu odhalilo plánované CT mozku před stereobiopsií progresi velikosti ložiska a perifokálního edému a bylo vysloveno podezření na absces. Ten byl následně ošetřen stereotaktickou aspirací s ponecháním drenáže v abscesové dutině. Pro výraznou hemoragickou příměs v drénu bylo indikováno kontrolní CT mozku, které potvrdilo krvácení do mozkového parenchymu, což si vyžádalo operační revizi. Po výkonu vznikla těžká hemiparéza vlevo, která postupně regredovala a po roce byla pouze lehká s akcentací na levé horní končetině akrálně. Na předoperačním rentgenovém snímku plic byla patrná čtyři drobná okrouhlá ložiska bazálně vpravo, se kterými byla pacientka předána do péče pneumologa.
Subjektivně se ale pacientka cítila stále unavená. Kontrolní MR mozku neprokazovalo recidivu abscesu. Pacientka byla dále v péči neurologa se zavedenou antiepileptickou terapií a v péči pneumologa pro nález patologického rozšíření venózních struktur na bázích oboustranně na CT plic. Pro zhoršující se hypoxemii s postupnou progresí stavu vyžadujícího domácí respirátor byla za rok po operaci mozkového abscesu indikována digitální subtrakční angiografie (DSA) plicního řečiště, která potvrdila AVM oboustranně bazálně. Ty byly ošetřeny embolizací. Po výkonu došlo k úpravě respiračních funkcí a vymizení hypoxemie (obr. 1).
Kazuistika 2
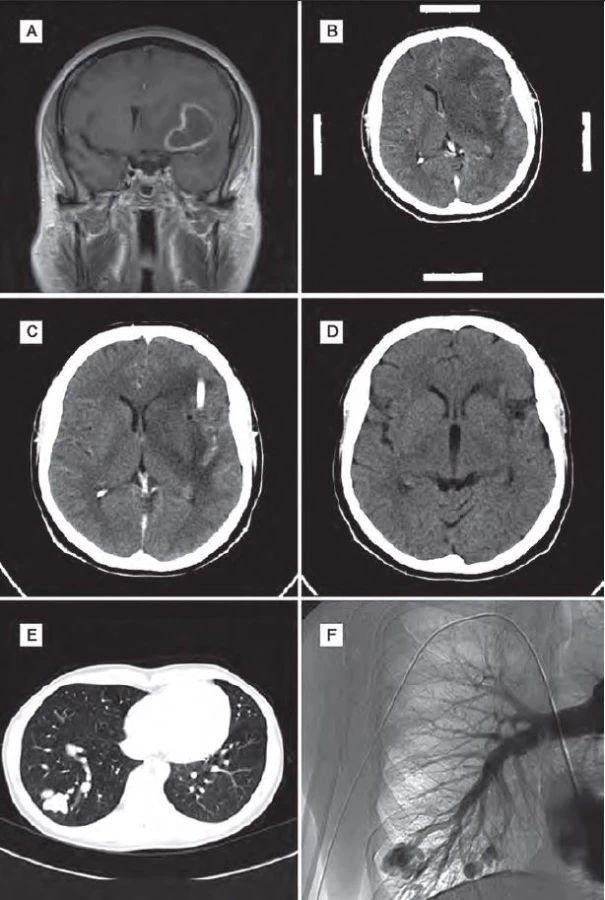
Žena (60 let) v minulosti absolvovala opakovaně leptání pro krvácení z nosu. Kromě hypertenze neměla žádné jiné významnější komorbidity. Rodinná anamnéza ve smyslu HHT byla negativní. Na základě náhodného nálezu na rentgenovém snímku plic bylo provedeno CT plic s nálezem plicní AVM. Embolizace AVM nebyla doporučena. Po roce pro náhle vzniklou cefaleu, zmatenost a pravostrannou hemiparézu byla pacientka hospitalizována na neurologii. Na CT a následně na MR mozku byla zjištěna expanze frontotemporoparietálně vlevo. Pro podezření na absces byla indikována stereotaktická punkce, aspirace a drenáž. Drén byl použit k opakované laváži abscesové dutiny po dobu tří dnů. Na kontrolním CT mozku byla výrazná regrese nálezu. Po stabilizaci stavu byla provedena DSA plic a embolizace zkratu. Cílená antibiotická terapie byla ponechána tři měsíce. Kontrolní CT mozku půl roku po operaci bylo bez známek recidivy abscesu. Vzhledem k negativnímu nálezu na CT a výrazně horší dostupnosti MR v minulosti nebylo toto vyšetření provedeno. Klinický stav pacientky se výrazně zlepšil, reziduálně zůstal organický psychosyndrom lehkého stupně a diskrétní pravostranná hemiparéza (obr. 2).
Kazuistika 3
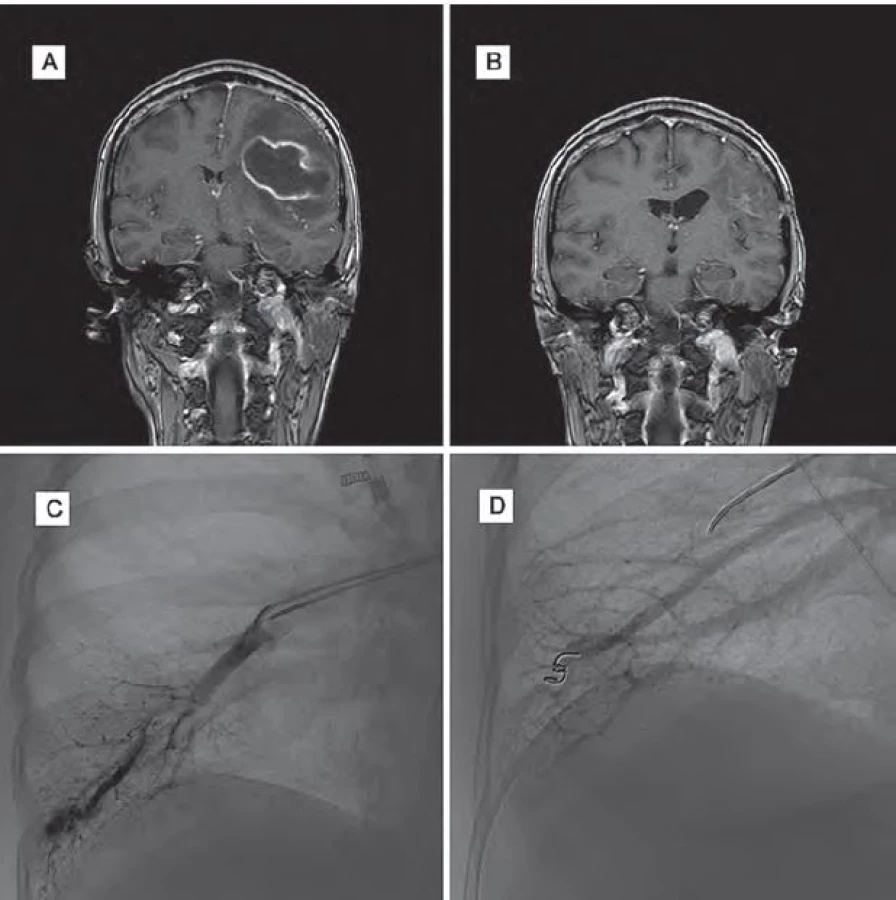
Žena (39 let) byla přijata do nemocnice pro cefaleu, rozvoj expresivní fatické poruchy, pravostranné hemiparézy a poruchy zraku. Její matka a sestra jsou sledovány pro HHT. Na MR mozku byl potvrzen absces frontoparietálně vlevo a pacientka podstoupila stereotaktickou aspiraci abscesu. Po výkonu se klinický stav pacientky zlepšil, zlepšila se fatická porucha i hybnost pravostranných končetin. Kontrolní MR mozku prokázala výraznou regresi velikosti abscesu. Na DSA se zobrazily oboustranné plicní AVM, které byly po stabilizaci stavu pacientky embolizovány. Po výkonu byla pacientka propuštěna do domácího ošetření se zavedenou cílenou antibiotickou terapií dle citlivosti na tři měsíce.
Klinický stav pacientky se výrazně zlepšil, momentálně přetrvává jen frustní oslabení pravostranných končetin a lehká porucha řeči (obr. 3).
Diskuze
Klinické projevy HHT jsou následkem abnormální vaskulární struktury [2]. Podle Bravemana et al je nejčasnějším stadiem detekovatelné léze HHT fokální dilatace postkapilárních venul, která progreduje, a postupně dochází ke spojení s dilatovanými arteriolami prostřednictvím kapilár, které postupně vymizí utvořením drobné arteriovenózní fistuly [9].
Vznik HHT je asociován s různými geny a představuje geneticky heterogenní skupinu [10]. Mutace genu kódujícího ENG na chromozomu 9q33– q34.1 je odpovědná za HHT prvního typu, zatímco mutace ACVRL1 genu na chromozomu 12q11– q14 se vyskytuje u HHT druhého typu [11]. Nepřítomnost těchto mutací u dalších rodin trpících HHT vedla k identifikaci třetího typu HHT s mutací na chromozomu 5q31.3– q32 [12] a čtvrtého typu HHT s mutací na chromozomu 7p14 [13]. Kromě toho byly objeveny další mutace v oblasti MADH4/ SMAD4 na chromozomu 18q21.2, které zapříčiňují vznik HHT a juvenilní polypózy [14], a v oblasti BMPRII, které zapříčiňují vznik HHT a plicní hypertenze [15]. Navíc bylo zjištěno, že vaskulární endoteliální růstový faktor (VEGF) jako signální protein podílející se na angiogenezi je zvýšen u pacientů s HHT [16]. Ovlivnění jeho exprese pak může mít vliv na fenotyp HHT. VEGF je zvýšen nejen v plazmě, ale také v nosní sliznici bez závislosti na vzniku a tíži epistaxe [17]. Závažnost klinických projevů HHT nekoreluje se žádnou specifickou mutací.
Ve 100 % případů se HHT manifestuje do 40 let věku. Iniciální diagnóza je určena na základě klinického vyšetření, osobní a rodinné anamnézy [1]. Často se objevující epistaxe u HHT se ale vyskytuje u řady dalších onemocnění a limituje stanovení diagnózy HHT. Podobně obtížná je diagnóza HHT v dětském věku vzhledem ke krvácení, které bývá relativně častým problémem u dětí. Nutné je samozřejmě vyloučit hematologickou příčinu krvácivých stavů. Onemocnění tak bývá často nediagnostikováno [18] a podobnou situaci lze předpokládat také v České republice, i když přesné údaje nejsou k dispozici.
Vzhledem k výrazné genetické heterogenitě HHT je možno detekovat mutace pouze u 70– 90 % nemocných [19]. Je tedy nezbytné zjistit konkrétní typ mutace u jedince postiženého HHT a teprve pak lze provést vyšetření u ostatních členů rodiny.
U pacientů s HHT nebo s podezřením na HHT se doporučuje provést kontrastní echokardiografii ke zjištění případných intrapulmonálních zkratů a následně provést CT kontrastní vyšetření plic. Kromě toho je nutno doplnit MR mozku k vyloučení AVM v této lokalizaci a auskultaci pro šelest v oblasti jater [2].
Léčba HHT je zaměřena na lokální a systémové symptomy, sledování lézí a prevenci komplikací asociovaných s AVM. Zatímco teleangiektázie se manifestují recidivujícím krvácením, u AVM komplikace vznikají jako následek zkratu, trombózy nebo embolu [20].
Epistaxe se objevují průměrně ve 12 letech s průměrnou frekvencí 18 epizod měsíčně [21]. Těžká epistaxe může zapříčinit vznik chronické anémie. Léčba spočívá v lokálním ošetření a medikamentózní terapii (tranexamová kyselina nebo estrogen‑ progesteronové preparáty), popřípadě léčbě anémie. Kožní teleangiektázie je možné ošetřit laserem z estetických důvodů nebo pro bolest. Teleangiektázie v gastrointestinálním traktu se léčí jejich koagulací s využitím endoskopické techniky.
Plicní AVM se častěji vyskytují u prvního typu HHT (v 75 %) než u druhého typu (44 %) [22]. U přibližně 30– 40 % pacientů s plicními AVM se můžeme setkat s trombotickými a embolickými příhodami vedoucími k iktu nebo mozkovému abscesu. Proto se doporučuje u plicních AVM jejich embolizace, pokud jsou zásobující cévy většího kalibru než 3 mm [23], a u menších lézí jejich observace, protože se mohou časem zvětšovat [24]. Antibiotická profylaxe je doporučována u každého pacienta s přítomností intrapulmonálního zkratu podstupujícího zubní nebo chirurgický zákrok. V případě difuzního postižení plicního parenchymu u těžce hypoxických pacientů je pak nezbytná transplantace plic.
Mozkové AVM se vyskytují častěji u pacientů s HHT prvního typu (15– 20 %) než u druhého typu (1– 2 %). Projevit se mohou v jakémkoli věku epileptickým záchvatem, bolestmi hlavy nebo intracerebrálním krvácením [25]. Spinální AVM se vyskytují pouze u 1 % pacientů s HHT a nejčastěji se klinicky manifestují progresivní myelopatií, radikulárními nebo sfinkterovými potížemi [26].
Výskyt vaskulárních hepatálních lézí není přesně znám, ale odhaduje se až u 74 % nemocných s HHT [27]. Většinou jsou klinicky asymptomatické, ale mohou se manifestovat srdečním selháním, biliárními chorobami nebo encefalopatií [27]. Asymptomatické léze se neléčí, protože nezpůsobují závažné komplikace. Jejich embolizace je riziková pro vznik infarktových ložisek. U rozsáhlého postižení pak bývá indikována transplantace jater [28].
I když diagnostická Curaçao kritéria splňovala pouze jedna pacientka, byli pro závažný nález řádně došetřováni i další dva pacienti pro pravděpodobnou diagnózu podle těchto kritérií. U všech pacientů byl chirurgicky ošetřen absces za aplikace celkově podávaných antibiotik a ošetřeny plicní AVM. Neprovedení embolizace plicní AVM v druhé kazuistice zapříčinilo komplikaci vznikem již zmiňovaného abscesu mozku a trvalých následků.
Závěr
HHT je relativně vzácné onemocnění, ale může způsobit závažné komplikace. U každého pacienta s mozkovým abscesem je nezbytné na toto onemocnění pomýšlet a v případě podezření je nutné jej řádně vyšetřit. V případě verifikované plicní AVM je třeba ji ošetřit a zabránit tak vzniku recidivujících mozkových abscesů. Péče o pacienty s HHT je komplexní a multioborová. Při průkazu HHT u jedince je také nutno vyšetřit rodinné příslušníky.
Použité zkratky
ACVRL1 aktivin receptoru podobná kináza 1 (Activin A Receptor Type II‑like Kinase 1)
AVM arteriovenózní malformace (Arteriovenous Malformation)
CT výpočetní tomografie (Computed Tomography)
DSA digitální subtrakční angiografie (Digital Subtraction Angiography)
ENG endoglin
HHT hereditární hemoragická teleangiektázie (Hereditary Hemorrhagic Telangiectasia)
MR magnetická rezonance (Magnetic Resonance)
TGFb transformující růstový faktor beta (Transforming Growth Factor Beta)
VEGF vaskulární endoteliální růstový faktor (Vascular Endothelial Growth Factor)
Podpořeno projektem (Ministerstva zdravotnictví) koncepčního rozvoje výzkumné organizace 65269705 (FN Brno).
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Přijato k recenzi: 24. 11. 2014
Přijato do tisku: 9. 3. 2015
MUDr. George Hanoun
Neurochirurgická klinika
LF MU a FN Brno
Jihlavská 20
625 00 Brno
e-mail: ghanoun@post.cz
Sources
1. Shovlin CL, Guttmacher AE, Buscarini E, Faughnan ME, Hyland RH, Westermann CJ et al. Diagnostic criteria for hereditary hemorrhagic teleangiectasia (Rendu‑ Osler‑ Weber syndrome). Am J Med Genet 2000; 91(1): 66– 67.
2. Sharathkumar AA, Shapiro A. Hereditary hemorrhagic telangiectasia. Haemophilia 2008; 14(6): 1269– 1280. doi: 10.1111/ j.1365‑ 2516.2008.01774.x.
3. Plauchu H, de Chadarevian JP, Bideau A, Robert JM. Age‑related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am J Med Genet 1989; 32(3): 291– 297.
4. Porteous ME, Burn J, Proctor SJ. Hereditary hemorrhagic telangiectasia: a clinical analysis. J Med Genet 1992; 29(8): 527– 530.
5. Kjeldsen AD, Vase P, Green A. Hereditary hemorrhagic telangiectasia: a population‑based study of prevalence and mortality in Danish patients. J Intern Med 1999; 245(1): 31– 39.
6. Römer W, Burk M, Schneider W. Hereditary hemorrhagic telangiectasia (Osler‘s disease). Dtsch Med Wochenschr 1992; 117(17): 669– 675.
7. Koubaa M, Lahiani D, Mâaloul I, Fourati H, Chaari L, Marrakchi Ch et al. Actinomycotic brain abscess as the first clinical manifestation of hereditary hemorrhagic telangiectasia – case report and review of the literature. Ann Hematol 2013; 92(8): 1141– 1143. doi: 10.1007/ s00277‑ 012‑ 1666‑ 0.
8. Govani FS, Shovlin CL. Hereditary hemorrhagic telangiectasia: a clinical and scientific review. Eur J Hum Genet 2009; 17(7): 860– 871. doi: 10.1038/ ejhg.2009.35.
9. Braverman IM, Keh A, Jacobson BS. Ultrastructure and three‑ dimensional organization of the telangiectases of hereditary hemorrhagic telangiectasia. J Invest Dermatol 1990; 95(4): 422– 427.
10. McAllister KA, Lennon F, Bowles‑ Biesecker B, McKinnon WC, Helmbold EA, Markel DS et al. Genetic heterogeneity in hereditary hemorrhagic telangiectasia: possible correlation with clinical phenotype. J Med Genet 1994; 31(12): 927– 932.
11. McDonald MT, Papenberg KA, Ghosh S, Glatfelter AA,Biesecker BB, Helmbold EA et al. A disease locus for hereditary hemorrhagic telangiectasia maps to chromosome 9q33– 34. Nat Genet 1994; 6(2): 197– 204.
12. Cole SG, Begbie ME, Wallace GM, Shovlin CL. A new locus for hereditary hemorrhagic telangiectasia (HHT3) maps to chromosome 5. J Med Genet 2005; 42(7): 577– 582.
13. Bayrak‑ Toydemir P, McDonald J, Akarsu N, Toydemir RM,Calderon F, Tuncali T et al. A fourth locus for hereditary hemorrhagic telangiectasia maps to chromosome 7. Am J Med Genet A 2006; 140(20): 2155– 2162.
14. Gallione CJ, Repetto GM, Legius E, Rustgi AK, Schelley SL, Tejpar S et al. A combined syndrome of juvenile polyposis and hereditary hemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet 2004; 363(9412): 852– 859.
15. Trembath RC, Thomson JR, Machado RD, Morgan NV, Atkinson C, Winship I et al. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2001; 345(5): 325– 334.
16. Cirulli A, Liso A, D‘Ovidio F, Mestice A, Pasculli G, Gallitelli M et al. Vascular endothelial growth factor serum levels are elevated in patients with hereditary hemorrhagic telangiectasia. Acta Haematol 2003; 110(1): 29– 32.
17. Sadick H, Naim R, Sadick M, Hörmann K, Riedel F. Plasma level and tissue expression of angiogenic factors in patients with hereditary hemorrhagic telangiectasia. Int J Mol Med 2005; 15(4): 591– 596.
18. Grosse SD, Boulet SL, Grant AM, Hulihan MM, Faughnan ME. The use of US health insurance data for surveillance of rare disorders: hereditary hemorrhagic telangiectasia. Genet Med 2014; 16(1): 33– 39. doi: 10.1038/ gim.2013.66.
19. Olivieri C, Pagella F, Semino L, Lanzarini L, Valacca C, Pilotto A et al. Analysis of ENG and ACVRL1 genes in 137 HHT Italian families identifies 76 different mutations (24 novel). Comparison with other European studies. J Hum Genet 2007; 52(10): 820– 829.
20. Kjeldsen AD, Oxhoj H, Andersen PE, Elle B, Jacobsen JP, Vase P. Pulmonary arteriovenous malformations: screening procedures and pulmonary angiography in patients with hereditary hemorrhagic telangiectasia. Chest 1999; 116(2): 432– 439.
21. Assar O, Friedman CM, White RI jr. The natural history of epistaxis in hereditary hemorrhagic telangiectasia. Laryngoscope 1991; 101(9): 977– 980.
22. Sabba C, Pasculli G, Lenato GM, Suppressa P, Lastella P, Memeo M et al. Hereditary hemorrhagic telangiectasia: clinical features in ENG and ALK1 mutation carriers. J Thromb Haemost 2007; 5(6): 1149– 1157.
23. White RI jr, Pollak JS, Wirth JA. Pulmonary arteriovenous malformations: diagnosis and transcatheter embolotherapy. J Vasc Interv Radiol 1996; 7(6): 787– 804.
24. Shovlin CL, Letarte M. Hereditary hemorrhagic telangiectasia and pulmonary arteriovenous malformations: issues in clinical management and review of pathogenic mechanisms. Thorax 1999; 54(8): 714– 729.
25. Fulbright RK, Chaloupka JC, Putman CM, Sze GK, Merriam MM, Lee GK et al. MR of hereditary hemorrhagic telangiectasia: prevalence and spectrum of cerebrovascular malformations. AJNR Am J Neuroradiol 1998; 19(3): 477– 484.
26. Cullen S, Alvarez H, Rodesch G, Lasjaunias P. Spinal arteriovenous shunts presenting before 2 years of age: analysis of 13 cases. Childs Nerv Syst 2006; 22(9): 1103– 1110.
27. Memeo M, Stabile Ianora AA, Scardapane A, Buonamico P, Sabba C, Angelelli G. Hepatic involvement in hereditary hemorrhagic telangiectasia: CT findings. Abdom Imaging 2004; 29(2): 211– 220.
28. Hillert C, Broering DC, Gundlach M, Knoefel WT, Izbicki JR, Rogiers X. Hepatic involvement in hereditary hemorrhagic telangiectasia: an unusual indication for liver transplantation. Liver Transpl 2001; 7(3): 266– 268.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2015 Issue 3
Most read in this issue
- Addenbrookský kognitivní test – orientační normy pro českou populaci
- Míšní šok – od patofyziologie ke klinickým projevům
- Diagnostika epileptických záchvatů
- Vzduchová embolie mozku – kazuistika