
Wilsonova nemoc
Wilson Disease
The Wilson disease is a rare hereditary metabolic disease with autosomal recessive inheritance. It is caused by a dysfunction of the ATP7B protein that hampers copper excretion into the bile ducts and its integration into the ceruloplasmin. Disease symptoms arise as a consequence of excess copper accumulation in various organs, especially in the liver and brain. Manifestations of the Wilson disease may be very diverse and it is necessary to consider this diagnosis in any patient aged 5– 50 years with extrapyramidal, cerebellar or psychiatric symptoms, especially in a setting of liver dysfunction. The most common neuropsychiatric symptoms include tremor, dysarthria, anxiety, depression, Parkinsonism, personality disorders, ataxia and dystonia. Final diagnosis requires the use of several auxiliary investigations: ophthalmic, genetic, brain magnetic resonance imaging and biochemical proof of disturbed copper metabolism. Early diagnosis and treatment is necessary to avoid irreversible brain and liver damage. The Wilson disease has an excellent prognosis subject to early therapy initiation and life- long regular monitoring of its effect.
Key words:
Wilson disease – neurologic manifestation – hepatolenticular degeneration – ATP7B mutation – chelation therapy
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
:
P. Dušek 1; R. Brůha 2; Andrea Burgetová 3
![]() ; D. Záhoráková 4; E. Růžička 1
; D. Záhoráková 4; E. Růžička 1
:
1. LF UK a VFN v Praze
Neurologická klinika a Centrum klinických neurověd
1; 1. LF UK a VFN v Praze
IV. interní klinika
2; 1. LF UK a VFN v Praze
Radiodiagnostická klinika
3; 1. LF UK a VFN v Praze
Klinika dětského a dorostového lékařství, Laboratoř pro studium mitochondriálních poruch
4
:
Cesk Slov Neurol N 2013; 76/109(5): 539-549
:
Minimonography
Práce byla podpořena grantem Univerzity Karlovy PRVOUK-P26/LF1/4.
Wilsonova nemoc je vzácné hereditární metabolické onemocnění s autozomálně recesivní dědičností. Příčinou je dysfunkce proteinu ATP7B způsobující poruchu vylučování mědi z hepatocytů do žlučových cest a inkorporaci mědi do ceruloplazminu. Příznaky onemocnění jsou způsobeny akumulací mědi v různých orgánech, zejména v játrech a mozku. Projevy onemocnění mohou být velmi různorodé a je třeba na něj myslet u pacientů s jakýmkoliv extrapyramidovým, cerebelárním či psychiatrickým postižením, zejména ve věku 5– 50 let, a při anamnéze hepatopatie. Nejčastějšími neuropsychiatrickými příznaky jsou třes, dysartrie, anxiózně‑depresivní syndrom, dále parkinsonský syndrom, poruchy osobnosti, ataxie a dystonie. K potvrzení diagnózy je třeba provést řadu pomocných vyšetření: oftalmologické, genetické, magnetickou rezonanci mozku a biochemický průkaz poruchy metabolizmu mědi. Stanovit diagnózu a zahájit terapii je nutné co nejdříve, neboť hrozí riziko ireverzibilního poškození mozku a jater. Onemocnění má výbornou prognózu v případě časně zahájené celoživotní terapie a pravidelného monitorování jejího efektu.
Klíčová slova:
Wilsonova nemoc – neurologická manifestace – hepatolentikulární degenerace – mutace ATP7B – helatační léčba
Úvod
Wilsonova nemoc (WN), dříve označovaná jako hepatolentikulární degenerace, je autozomálně recesivně dědičné onemocnění, které je způsobeno poruchami genu pro ATP7B protein uloženého na dlouhém raménku 13. chromozomu [1,2]. Nejčastěji uváděná hodnota prevalence WN je 1 : 30 000, extrémní odhady ale kolísají mezi 1 : 7 000 až 1 : 100 000 [3]. Dle epidemiologických údajů lze tedy předpokládat, že v České republice je cca 300 pacientů s WN, z nichž se přibližně u poloviny iniciálně manifestují neuropsychiatrické příznaky [4– 6].
Patofyziologie
Příčinou klinických projevů WN je porucha metabolizmu mědi, jejíž homeostáza je za fyziologických okolností udržována biliární exkrecí za pomoci proteinu ATP7B. ATP7B je protein s ATPázovou aktivitou, který je exprimován zejména v hepatocytech. Intracelulární distribuce ATP7B je závislá na koncentraci mědi v hepatocytu. Při její normální koncentraci je ATP7B lokalizován v Golgiho aparátu a podílí se na inkorporaci mědi do kuproproteinů, zvláště ceruloplazminu. Při její zvýšené koncentraci dochází k přesunu ATP7B do buněčné membrány žlučového pólu hepatocytu, kde zajišťuje exkreci mědi do žlučových kanálků. V důsledku dysfunkce ATP7B je u WN tedy omezeno vylučování mědi do žlučových cest a inkorporace mědi do ceruloplazminu [7]. Postupná akumulace mědi v hepatocytech vede k jejich dysfunkci a v konečném důsledku k nekróze. V průběhu progrese nemoci dochází k vyčerpání kapacity jaterních buněk pro skladování mědi a ta se uvolňuje do krevního řečiště a ukládá se v tkáních těla, zejména v mozku, rohovce a ledvinách, ale i v jiných orgánech. Předpokládá se, že ukládaná měď je pro tkáně toxická zvláště vinou zvýšení tvorby volných kyslíkových radikálů. To vede k oxidativnímu stresu [8], ale pravděpodobně působí i dalšími mechanizmy, jako například indukcí apoptózy [9]. Na intracelulární úrovni dochází zejména k postižení mitochondrií a dysfunkci dýchacího řetězce [10].
Klinický obraz
Příznaky WN se mohou objevit v kterémkoliv věku, ale ve většině případů začíná onemocnění mezi 5. a 40. rokem. U 3 % nemocných se příznaky objevují až po 40. roce a byly popsány případy začátku onemocnění před 3. rokem i po 70. roce života [11,12]. Klinické projevy onemocnění jsou velmi variabilní, nejčastější a nejnápadnější jsou příznaky jaterní a neuropsychiatrické. Arbitrárně se někdy rozlišuje hepatální, neurologická, psychiatrická a smíšená forma. Zhruba 50 % pacientů se manifestuje neurologickými příznaky a 50 % jaterními příznaky, přičemž cca 60 % pacientů s neurologickou formou má již v době diagnózy těžké postižení jater dosahující stupně cirhózy a tato skupina tvoří tzv. smíšenou formu [6]. Časté jsou rovněž příznaky oftalmologické (Kayser- Fleischerův prstenec, slunečnicová katarakta), renální (urolitiáza, hematurie, hyperkalciurie, nefrokalcinóza), kostní (chondrokalcióza, osteoartritis) a hematologické (akutní hemolytická anémie, trombocytopenie). Vzácněji se WN může projevit neplodností a opakovanými potraty, symptomy kardiálními, pankreatitidou a poruchou funkce příštítných tělísek [13,14].
V neurologické praxi je třeba na diagnózu WN myslet u pacienta s jakýmkoliv psychiatrickým, extrapyramidovým či cerebelárním příznakem, který nemá jiné vysvětlení. Je třeba upozornit na to, že u pacienta s pokročilým hepatálním selháním v rámci nerozpoznané WN mohou být neurologické příznaky modifikovány a někdy mylně přičítány výlučně jaterní encefalopatii.
Neuropsychiatrické příznaky
První příznaky se u neuropsychiatrické formy projevují obvykle ve 2.– 3. dekádě, tedy později než u čistě hepatální formy, která typicky začíná již v dětství [15]. Frustní poruchy jsou však často detekovatelné mnohem dříve, dynamika jejich progrese navíc může být různá a mnohdy nebývá lineární. U dětí jsou někdy pozorovány dlouhodobě stacionární frustní preklinické projevy behaviorální (únavnost, podrážděnost, zhoršení školní výkonnosti) a neurologické (nešikovnost, zhoršení rukopisu či jemný tremor). V adolescenci nebo časné dospělosti může dojít k prudké progresi těchto příznaků, která během několika měsíců způsobí těžkou invaliditu.
Neurologické projevy bývají velmi pestré, mezi nejčastější patří třes a dysartrie a dále různé kombinace parkinsonského syndromu, dystonie, cerebelární ataxie a instability chůze. Porucha hybnosti je někdy bizarní a může evokovat dojem psychogenní poruchy. Diferenciální diagnóza WN je velmi široká a onemocnění se může projevit jakýmkoliv extrapyramidovým, cerebelárním či psychiatrickým příznakem (tab. 1).
Někteří autoři rozdělují neurologické projevy WN do čtyř typů:
- parkinsonský syndrom,
- pseudoskleróza s dominujícím třesem a dysartrií,
- ataxie,
- dystonický syndrom [16].
Vzhledem k jejich variabilitě a častým kombinacím ale nelze neurologické příznaky v řadě případů do těchto kategorií jednoznačně zařadit.
Tremor může být jakéhokoliv typu: uni- i bilaterální, vyskytující se na hlavě či horních končetinách, klidový, statický nebo intenční. Fenotypově může připomínat akcentovaný fyziologický, esenciální, parkinsonský či rubrální tremor. Charakteristický, ale zdaleka ne vždy přítomný, je takzvaný „wingbeating“ tremor, hrubý nepravidelný proximální, asymetrický, avšak obvykle bilaterální třes rubrálního typu, který je nejlépe patrný při vyšetření horních končetin s abdukcí v ramenních kloubech a flexí v loktech a připomíná mávání křídel. Tremor by neměl být zaměňován za asterixis neboli negativní myoklonus, který se manifestuje krátkým výpadkem svalového tonu ruky v zápěstí při předpažených horních končetinách s dorziflexí v zápěstí. Tento příznak je projevem jaterní encefalopatie u WN a někdy bývá terminologicky ne zcela přesně nazýván „flapping tremor“.
Dysartrie může být důsledkem léze mozkového kmene, mozečku i bazálních ganglií, tedy struktur, které jsou u WN nejčastěji postiženy. To vysvětluje její vysokou prevalenci u WN (tab. 1). Dysartrie bývá většinou smíšená se spastickou, cerebelární, hypokinetickou a dystonickou komponentou. K dysartrii se velmi často přidružují další poruchy bulbárního svalstva – dysfagie, faryngeální dysmotilita a dále i postižení obličejového svalstva projevující se jako oro‑buko‑linguální dyskineze, blefarospazmus a grimasování způsobující typickou facies s retrahovanými rty, otevřenými ústy, sialoreou a tzv. prázdným úsměvem (vacuous smile). Někdy bývá tento výraz obličeje nepřesně uváděný jako risus sardonicus.

Dystonie bývá zpočátku fokální, kromě výše popsané obličejové dystonie se může manifestovat blefarospazmem, cervikální dystonií nebo grafospazmem. S progresí onemocnění dochází postupně i ke generalizaci dystonie, která v době před zavedením chelatační léčby obvykle ústila v imobilizaci a bolestivé kontraktury.
Parkinsonský syndrom obvykle nebývá tremor dominantní a spíše se projevuje akinezí, rigiditou a posturální instabilitou. U pacientů s parkinsonským syndromem byla prokázána korelace mezi klinickou tíží a pre‑ i postsynaptickým dopaminergním deficitem ve striatu zjištěným pomocí scintigrafie [17]. S tím může souviset i někdy částečně zachovaná pozitivní odpověď na dopaminergní terapii.
Spasticita s pozitivními pyramidovými iritačními příznaky se objevuje vzácněji, stejně jako cerebelární ataxie nebo chorea (tab. 1).
Prevalence epileptických záchvatů u WN bývá udávána kolem 6 %, většinou se objevují těsně po nasazení chelatační terapie a jejich prognóza bývá dobrá [18].
Paralelně s neurologickými se rozvíjejí i psychiatrické příznaky, které alemohou být i první manifestací nemoci. Pečlivá anamnéza může objevit iniciální psychiatrické příznaky dokonce až u 2/ 3 pa-cientů [19]. Nejčastěji se jedná o změny osobnosti, podrážděnost a úzkostně‑depresivní syndrom. Dalšími projevy mohou být únavnost, poruchy spánku, poruchy paměti, změny v sexuologické oblasti nebo i psychotické projevy charakteru poruchy s bludy nebo schizoafektivní poruchy [20]. Psychiatrická forma WN je obtížně diagnostikovatelná, a často tak dochází k pozdnímu rozpoznání choroby a oddálenému nasazení léčby. Varovnými příznaky WN v psychiatrické praxi jsou anamnéza hepatopatie, citlivost na neuroleptika a pozitivní rodinná anamnéza [21]. Psychiatrické příznaky navíc mnohdy přetrvávají i při dlouhodobé léčbě [22].
U jaterní encefalopatie, která může být důsledkem hepatopatie u WN, obvykle dominují kognitivní poruchy či poruchy vědomí, zejména zpomalení psychomotorického tempa, apatie, poruchy pozornosti a krátkodobé paměti, somnolence, dezorientace nebo zmatenost. Extrapyramidové projevy s výjimkou asterixis se u hepatální encefalopatie vyskytují vzácněji, nejčastěji se jedná o tremor či hypokineticko‑rigidní syndrom. Pro neurologické příznaky WN způsobené akumulací mědi platí opak: obvykle dominují extrapyramidové příznaky, zatímco kognitivní poruchy jsou pouze mírné. V nejasných případech může pomoci stanovení sérové hladiny amoniaku.
Hepatální příznaky
Projevy jaterního postižení jsou rovněž velmi variabilní. Může se projevit pouze asymptomaticky zvýšenými hodnotami jaterních enzymů nebo onemocněním připomínající virovou akutní hepatitidu se spontánním ústupem obtíží. Jedná se vždy o chronické jaterní onemocnění, které se také může manifestovat jako akutní jaterní selhání, často asociované s Coombs- negativní hemolytickou anémií, nápadným ikterem a renálním selháním. WN tedy patří do diferenciální diagnostiky akutního jaterního selhání u mladého pacienta, přičemž je příčinou 6– 12 % jaterních selhání, které jsou indikovány k akutní transplantaci jater [23]. K akutnímu jaternímu selhání může dojít i při náhlém vysazení chelatační léčby.
Jindy probíhá jaterní léze pod obrazem steatózy, steatohepatitidy či chronické hepatitidy s možným vývojem do stadia cirhózy. Dominujícími příznaky ve fázi pokročilé cirhózy může být nauzea, nechutenství, hubnutí a žloutenka, doprovázené ascitem či hemoragickou diatézou. Žloutenka může být způsobena také akutní hemolytickou anémií, která vzácně bývá také iniciálním příznakem WN [24]. Pacienti s neurologickou formou WN mají nezřídka v anamnéze údaj o elevaci jaterních testů či prodělané hepatitidě „nejasného původu“ předcházející neurologické postižení o řadu let a v době diagnózy mají již ve většině případů známky chronického jaterního postižení [12]. Neurologickou formu WN lze tedy chápat jako důsledek „benigně“ subklinicky probíhající akumulace mědi v játrech, díky čemuž není onemocnění včas rozpoznáno ani léčeno, a umožní se tak akumulace toxického množství mědi v mozku.
Oftalmologické příznaky
Nejtypičtějším oftalmologickým příznakem je Kayser- Fleischerův (KF) prstenec, který vzniká ukládáním mědi v descemetské membráně rohovky. Není zcela specifický pro WN, může být přítomen u chronických cholestatických onemocnění jater různé etiologie (např. u primární biliární cirhózy). Depozita mědi mají zlatěhnědé zbarvení a nejprve se objevují při horním okraji rohovky, v další fázi jsou patrné při dolním okraji rohovky a teprve posléze tvoří celý prstenec [25]. Nejlépe je vidět u lidí s modrou duhovkou, kdy bývá patrný i prostým okem, většinou je ale nutné k jeho potvrzení či vyloučení oftalmologické vyšetření štěrbinovou lampou. KF prstenec je přítomen u 80– 95 % pacientů s neurologickou formou WN, u jaterní formy a u asymptomatických nosičů mutace je jeho výskyt kolem 50 % [13].
Dalším specifickým, ale vzácnějším příznakem je slunečnicová katarakta, opacifikace v čočce způsobené ukládáním mědi v jejím pouzdře.
Intenzita KF prstence, stejně jako změny v čočce se snižují při farmakologické léčbě nebo po transplantaci jater a někdy mohou zcela vymizet.
Laboratorní nálezy
Laboratorní vyšetření při diagnostice WN spočívá zejména v průkazu abnormit metabolizmu mědi. Zvýšení jaterních transamináz je patrné u většiny pacientů s jaterní formou, ale často nebývá přítomno u pacientů s neuropsychiatrickou formou. Hodnoty transamináz navíc nekorelují se stupněm postižení jater. Pro diagnózu WN svědčí kombinace sníženého ceruloplazminu, vysoké volné mědi v séru, vysokého odpadu v moči a zvýšení obsahu mědi v jaterní sušině. Žádné vyšetření není dostatečně specifické samo o sobě (tab. 2), a vždy je proto nutné vyšetřit celý panel [13,26].
Ceruloplazmin je protein akutní fáze s feroxidázovou aktivitou, který je snížen u většiny nemocných s WN, včetně presymptomatického stadia. 5– 15 % pacientů s WN má ovšem pouze lehce subnormální nebo normální hladinu. Zejména v případě akutního jaterního selhání bývá hladina ceruloplazminu zvýšená a prediktivní hodnota vzhledem k diagnóze WN velmi nízká (tab. 2). Vyšetření ceruloplazminu má při zvolené hraniční hodnotě 0,2 g/ l senzitivitu 80– 95 %. Vzhledem k tomu, že ceruloplazmin je u WN zčásti enzymaticky neaktivní, je citlivější metodou enzymatické stanovení jeho feroxidázové aktivity v séru, které se ovšem rutinně neprovádějí [27]. Hladina ceruloplazminu v čase značně kolísá, a při nejasné diagnóze je tedy vhodné vyšetření zopakovat. Dle doporučení je velmi nízká hodnota (< 0,05 g/ l) silným prediktorem diagnózy WN, zatímco lehce abnormní hodnoty jsou nespecifické a vyžadují další vyšetřování [26].
Odpad mědi v moči za 24 hod je u pacientů s WN obvykle zvýšen nad 1,6 µmol/ 24 hod, ale již při hodnotě 1 či dokonce 0,6 µmol/ 24 hod se doporučuje další vyšetření [13]. Zejména děti a presymptomatičtí pacienti mají nejméně ve 20 % případů odpad mědi močí nižší než 1,6 µmol/ 24 hod. Pacienta je před sběrem moči nutno upozornit na dodržování diety s nízkým obsahem mědi (blíže v kapitole Terapie). U neléčených pacientů odráží odpad močí sérovou hladinu volné mědi. Odpady mědi močí se u WN zvýší po podání penicillaminu několikanásobně, čehož se diagnosticky využívá při penicilaminovém testu, který je validován pouze v dětské populaci. Při zvolené hranici 25 µmol/ 24 hod má penicilaminový test s podáním dvou dávek 500 mg v rozpětí 12 hod u symptomatických pacientů senzitivitu 92 % a specificitu 93 % [28].
Sérová hladina volné mědi je důležitější diagnostický ukazatel v porovnání s celkovou plazmatickou mědí, která odráží především měď navázanou na ceruloplazmin a u WN bývá snížená. Stanovení volné mědi je prováděno výpočtem, odečtením mědi vázané na ceruloplazmin od celkové hladiny mědi. Sérová hladina vyšší než 1,6 µmol/ l je považována za patologickou. Hladina mědi je mimo jiné závislá i na aktuálním příjmu potravou a abnormní výsledky u zdravých ani normální výsledky u WN nejsou výjimkou (tab. 2).
Jaterní biopsie a stanovení obsahu mědi v jaterní sušině je nejcitlivější a nejpřesnější diagnostický test WN. Hladina mědi u zdravých osob nepřekračuje 50 µg/ g (0,8 µmol/ g) sušiny, zatímco u WN, včetně presymtomatických jedinců, obvykle přesahuje 250 µg/ g. Při takto stanovené hranici má toto vyšetření senzitivitu 83,3 % a specificitu 98,6 % [29]. Hodnoty mezi 50 a 250 µg/ g jsou nespecifické, bývají nalézány u heterozygotů, ale i některých pacientů. Falešně negativní výsledek může být zapříčiněn nerovnoměrným uložením mědi v játrech, kdy jaterní biopsie nezíská dostatečně reprezentativní vzorek (tab. 2). Histochemická zkouška na měď rhodaninem nebo orceinem je pozitivní pouze u 10 % pacientů a není náhradou za kvantitativní stanovení mědi v sušině [13].
Genetika
V současné době je známo minimálně 600 mutací v ATP7B genu, z nichž téměř 500 je prokazatelně patogenních. Mutace se mohou vyskytnout ve kterémkoliv z 21 exonů tohoto genu, nebo v přilehlých nekódujících regulačních oblastech [30]. V ČR, stejně jako v celé střední a východní Evropě je zdaleka nejčastější mutace p.H1069Q ve 14. exonu, která má 50– 60% podíl na všech mutovaných alelách. Spolu s dalšími čtyřmi nejčastějšími mutacemi je odpovědná za 70 % případů z celkového počtu případů [31]. Další mutace jsou již velmi vzácné a při negativitě skríningovho vyšetření pěti prevalentních mutací je nutno provést sekvenaci celého genu. Pozitivní výsledek genetického vyšetření prakticky potvrzuje diagnózu WN.
Frekvence mutací v ATP7B genu v populaci není přesně známa, nejčastěji uváděná hodnota je 1 : 90, ale dle nových populačních studií by se mohlo jednat až o 1 : 40 [3]. Riziko onemocnění pro potomka pacienta s WN se tedy pohybuje v rozmezí 1 : 80– 1 : 180. Genetické vyšetření u rodinných příslušníků se rutinně doporučuje pouze pro sourozence a potomky pacienta s WN. Pokud je známa kauzální mutace, provádí se genetické vyšetření cílené pouze na její detekci.
Onemocnění se vyznačuje obrovskou klinickou heterogenitou, která nebyla doposud zcela vysvětlena. Bylo prokázáno, že část klinické variability je způsobeno typem mutace. U prevalentní mutace p.H1069Q zůstává protein aspoň částečně funkční, onemocnění se tak projeví relativně v pozdějším věku a nástup symptomů bývá pozvolnější. Tato mutace je spojována s vyšším podílem neuropsychiatrické formy WN [32]. Genetická variabilita však vysvětluje fenotypové rozdíly pouze z malé části a onemocnění se může projevit u pacientů s identickou mutací, včetně monozygotních dvojčat, zcela odlišně [33].
Nálezy v zobrazovacích vyšetřeních
Změny v zobrazení mozku magnetickou rezonancí (MR) mají téměř všichni pacienti s neurologickou formou WN a někteří pacienti s jaterní formou i v presymptomatickém stadiu [34]. Existují však i pacienti s WN, kteří mají neurologické příznaky a normální nález na MR [15]. Typickým obrazem jsou hyperintenzní změny v T2 vážených obrazech v oblasti striata, globus pallidus, pontu a mezencefala, méně často i v thalamech a mozečku. Ty jsou někdy doprovázené i hypointenzitami v T2 vážených obrazech, zejména v globus pallidus. Předpokládá se, že podkladem T2 hyperintenzit jsou glióza, nekróza a cystické změny spolu s lokálním edémem. Původ hypointenzit v T2 vážených obrazech není zcela vysvětlen, může se jednat o paramagnetický efekt depozit ferritinu. U pokročilé jaterní formy WN byly popsány hyperintenzity v T1 vážených obrazech, nejčastěji v globus pallidus, které je možné pozorovat i u pacientů s chronickým jaterním onemocněním jiné etiologie. Předpokládá se, že odpovídají depozitům manganu. Demyelinizační změny, někdy až připomínající roztroušenou sklerózu, bývají vzácně popisovány v bílé hmotě supratentoriálně. Jsou spojovány s rezistencí na terapii, psychiatrickými příznaky a epilepsií [35]. Velmi častým nálezem je mozková atrofie, zvláště kortikální, cerebelární a kmenová. Morfometrická studie prokázala nejvýraznější úbytek tkáně v oblasti kmene [36].
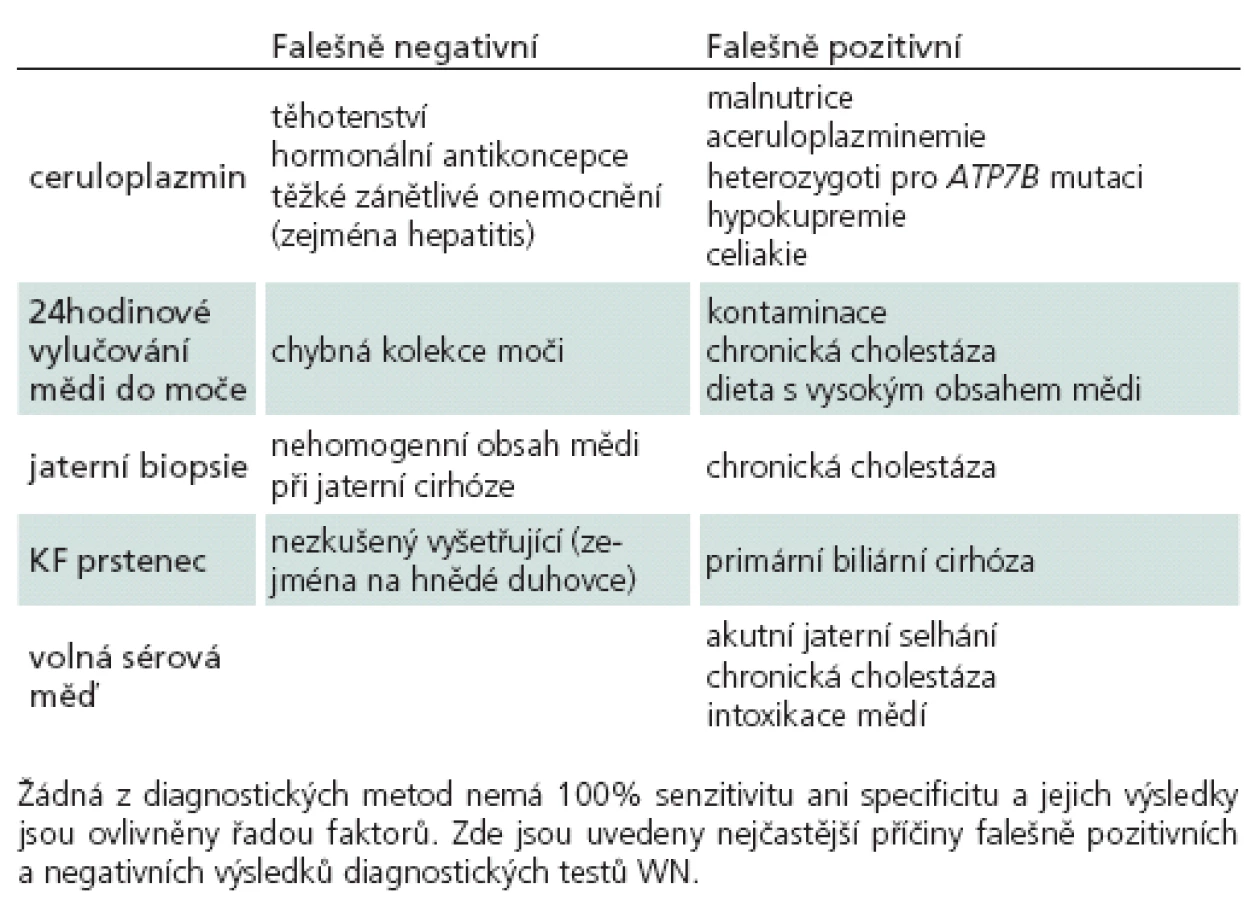
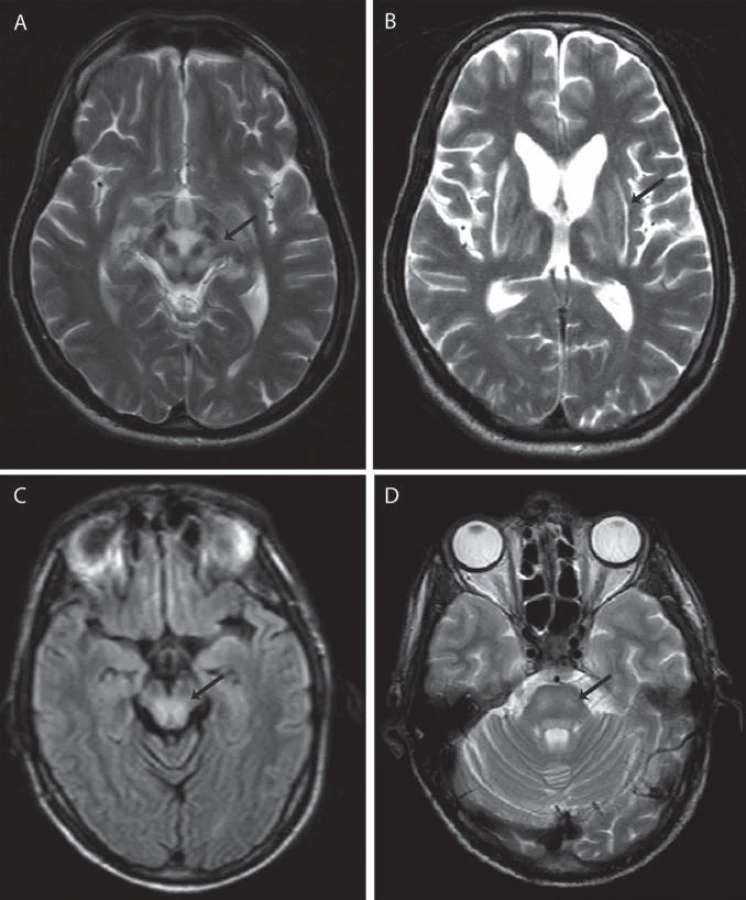
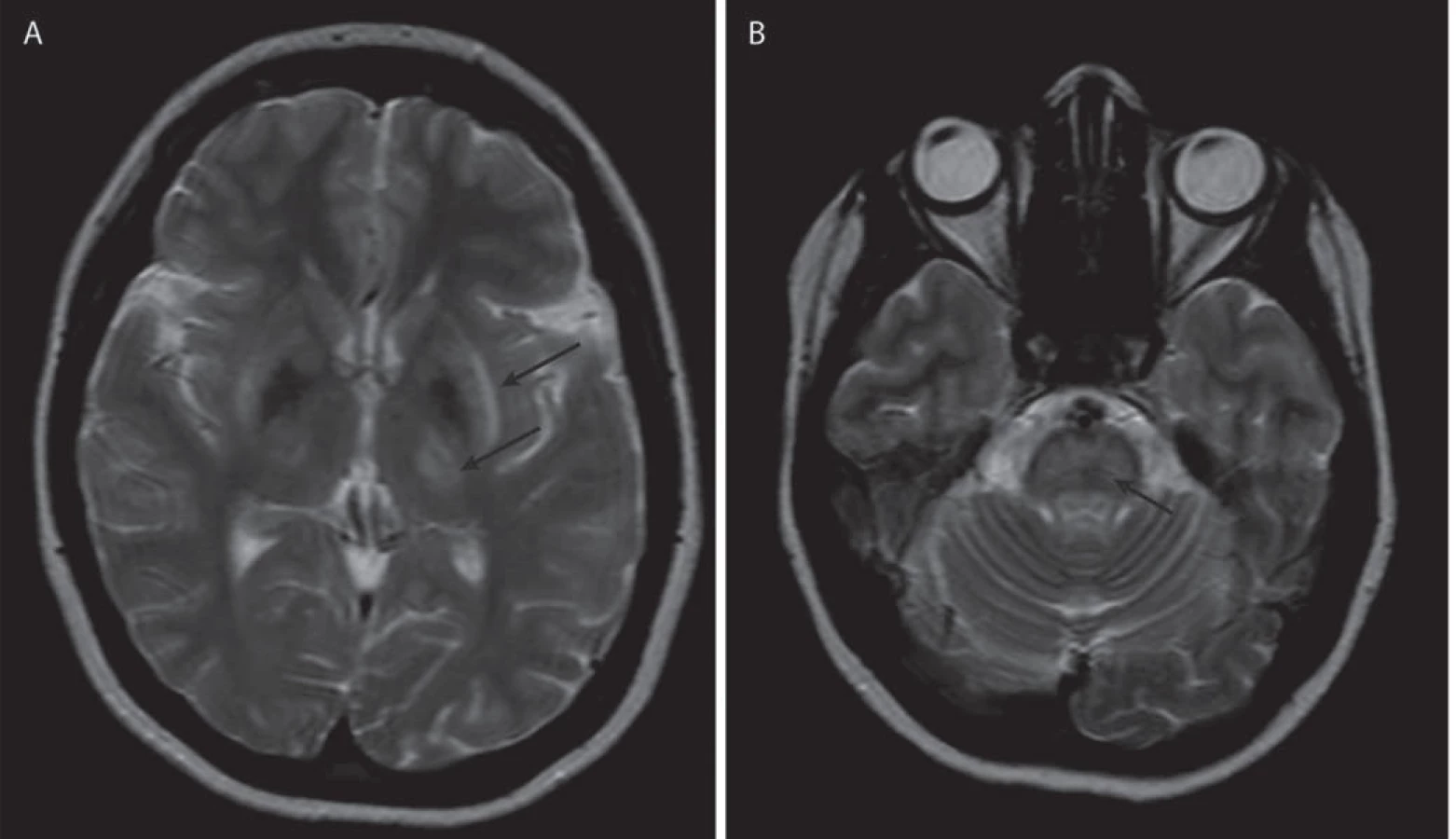
Jako specifický MR nález bývá uváděn obraz medvídka pandy a příznak jasného claustra v T2 vážených obrazech (obr. 1a, b), v rozsáhlé studii byly však nalezeny pouze u 12 %, resp. 4 % pacientů [34]. Dalšími téměř patognomickými nálezy jsou T2 hyperintenzní léze bílé hmoty v mezencefalickém tectu a v pontu (obr. 1c, d), kde mohou dosáhnout až obrazu centrální pontinní myelinolýzy. Ty se vyskytují až u 80 % pacientů [37]. Současná přítomnost změn signálu v kmeni, bazálních gangliích a thalamu je také velmi typická pro WN (obr. 2a, b). Patologické nálezy na MR ustupují při farmakologické léčbě i po transplantaci jater a mohou i zcela vymizet, atrofie však zůstává nezměněna [38]. Rozsáhlá atrofie a změny v bílé hmotě jsou prognosticky nepříznivé, ale bylo popsáno i jejich vymizení po dlouhodobé léčbě [39].
Transkraniální sonografie může detekovat hyperechogenitu v oblasti nucleus lentiformis a/nebo substantia nigra dokonce i v případě normálního nálezu na MR. Neuropatologický podklad tohoto nálezu však není jasný [40,41].
Diagnostický postup
Diagnostika WN se opírá o klinické, laboratorní, oftalmologické, radiologické a genetické nálezy. Ačkoliv je u typických případů diagnostika relativně jednoduchá, může být vzhledem k velké variabilitě klinických projevů a k časté nepřítomnosti některých charakteristických nálezů složitá. Průměrná latence od objevení se prvních příznaků k diagnóze je delší u neurologické v porovnání s hepatální formou, často kolem 24– 48 měsíců [15]. Vzhledem k tomu, že žádná z paraklinických metod nemá dostatečnou senzitivitu ani specificitu, je k definitivní diagnóze vždy třeba vzít v úvahu výsledky všech vyšetření. Pro diagnostické účely byla vyvinuta skórovací tabulka (tab. 3) bodující přítomnost či nepřítomnost patologických projevů. Bodový součet je uváděn jako Ferenciho či Leipzig skóre a vyjadřuje pravděpodobnost diagnózy WN [42]. Tento skórovací systém byl vyvinut převážně hepatology a není zcela praktický pro neurologické pacienty, neboť bodově nezohledňuje současnou přítomnost neurologického postižení a typického nálezu na MR vyšetření.
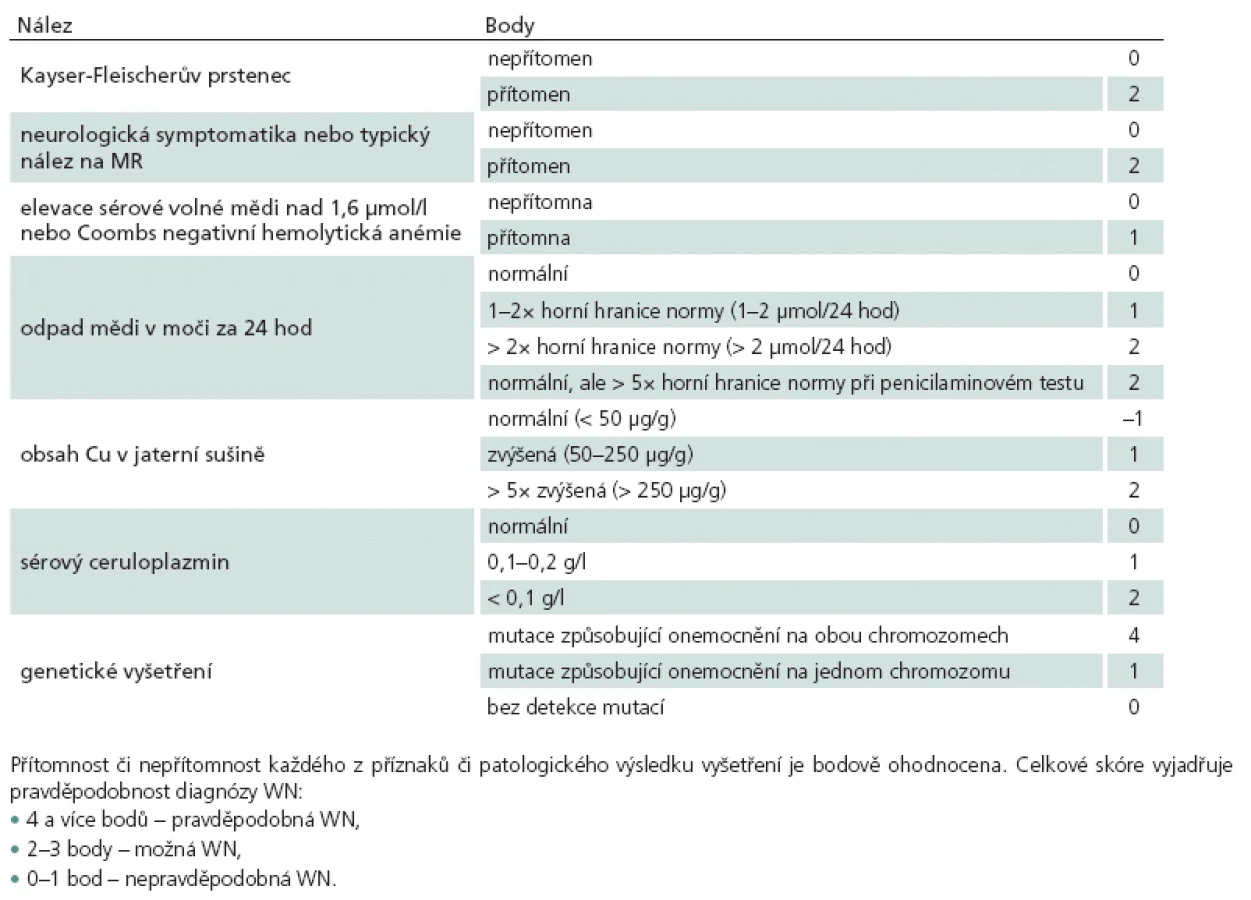
Doporučený postup při diagnostice pacienta s klinickým podezřením na neuropsychiatrickou formu WN je znázorněn na obr. 3. Doporučuje se nejprve provést vyšetření ceruloplazminu, oftalmologické vyšetření štěrbinovou lampou a stanovení odpadu mědi močí za 24 hod. V případě dostupnosti je vhodné doplnit i vyšetření volné sérové mědi. Dle výsledků a charakteru klinických obtíží pacienta je dále na místě doplnit MR mozku, genetické vyšetření a jaterní biopsii [26].
![Doporučený postup při vyšetření pacienta s podezřením na WN.
Upraveno dle [23], jedná se o obecné doporučení vycházející z naší klinické zkušenosti a v konkrétních případech lze postupovat jinak.
Typickými nálezy na MR v T2 vážených obrazech jsou: obraz medvídka pandy, příznak jasného claustra, hyperintenzity v mezencefalickém tectu a hyperintenzity v pontu připomínající osmotickou myelinolýzu.](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image/5ff35fe9da2953eed0bbae159e31562e.jpg)
Terapie
Léčba WN spočívá v nastolení negativní bilance mědi snížením přísunu nebo zvýšením exkrece pomocí chelátů. Princip chelatační terapie je vazba mědi na farmakum a její následné vyloučení. K dispozici jsou v současné době dva preparáty, penicilamin a trientin. Ve skupině 380 pacientů bylo po nasazení chelatační léčby pozorováno zlepšení hepatální symptomatiky v 90 % a neurologické symptomatiky v 55 % případů [43].
Důležitý aspekt léčby je i správná dieta. Pacienti by se měli vyhýbat játrům, čokoládě, ořechům, mořským plodům, houbám a luštěninám. Měď obsažená v zelenině má menší biologickou dostupnost, a vegetariánská dieta může tedy sama o sobě zmírnit pozitivní bilanci mědi [44]. Pozor je třeba dát i na obsah mědi ve vodě a užívání vitaminových doplňků. U žen je kontraindikováno používání hormonální antikoncepce obsahující estrogeny a nitroděložní tělíska obsahující měď. Snížit přísun mědi do organizmu lze také farmakologicky pomocí zinku a tetrathiomolybdenanu, které inhibují její absorpci ze střeva.
Penicilamin (dimetylcystein) váže měď s velkou afinitou, a je tedy velmi účinným chelátem, ale jeho užívání je spojeno s relativně vysokým výskytem nežádoucích účinků. Doporučovaná startovací dávka je 150 mg denně s postupným zvyšováním na 600 až 1 800 mg denně během 4– 8 týdnů. Spolu s penicilaminem by měl být užíván pyridoxin v dávce 20– 40 mg/ den. Relativně vzácnou, ale závažnou komplikací je akutní toxická reakce, která se projeví obvykle do dvou týdnů po zahájení terapie jako kožní exantém, lymfadenopatie, horečka, eozinofilie, leukopenie a trombocytopenie. Častou komplikací spojenou zejména s příliš rychlou eskalací dávky penicilaminu je zhoršení neurologické symptomatiky. Proto je doporučováno zvyšovat dávku pozvolna a monitorovat přitom klinické příznaky. Po několika letech chronického užívání penicilaminu se vzácně mohou objevit další komplikace, především lupus‑like syndrom s polyartritidou a pozitivitou antinukleárních protilátek, dále pemfigus a progerické změny kůže. Mezi raritní pozdní komplikace patří i myastenický syndrom, nefrotický syndrom, polymyositis, polyneuropatie, optická neuritis, trombocytopenie a IgA deficit.
Alternativa penicilaminu je trientin,který se obvykle používá v dávkách 900– 2 100 mg/ den. Mechanizmus účinku je podobný jako u penicilaminu, je však méně potentní ve vylučování mědi a při jeho užívání častěji perzistují neurologické příznaky. Jeho výhodou je naopak menší riziko nežádoucích účinků, proto se používá u pacientů netolerujících penicilamin [43].
Zinek působí nepřímo, indukuje tvorbu metalothioneinu, který pak v enterocytech váže měď a nedovolí její vstřebání. Je dostupný ve formě sulfátu a acetátu a doporučená dávka je 3krát denně 50 mg ekvivalentu elementárního zinku. Z nežádoucích účinků se mohou objevit sideropenická anémie a zejména gastrointestinální obtíže, které jsou v případě sulfátu velmi časté. Zinek ve formě acetátu je lépe tolerován. Má poměrně pomalý nástup účinku a vytvořená negativní bilance mědi není příliš velká. Proto se obvykle nepoužívá jako terapie první volby u symptomatických pacientů, zvláště s hepatální formou WN. Někteří autoři doporučují jeho iniciální nasazení u neurologické formy WN [13]. Zinek je ideální volba pro presymptomatické pacienty a bývá používán také při udržovací terapii a v těhotenství.
Tetrathiomolybdenát tvoří s mědí a albuminem komplexy ve střevě, a zabraňuje tak jejímu vstřebávání. Tyto komplexy se navíc tvoří i v krvi, kde neutralizují toxickou volnou měď. V současné době se jedná o experimentální terapii, která není komerčně dostupná [44].
V neposlední řadě patří do léčby i jaterní transplantace, jež je léčebnou metodou volby u fulminantního jaterního selhání. Výjimečně se může transplantace indikovat i pro léčbu dekompenzované jaterní cirhózy. V některých případech měla jaterní transplantace velmi dobrý efekt také na neurologickou a psychiatrickou symptomatiku [45].
Monitorování terapie a dlouhodobá prognóza
Předpokládá se, že onemocnění má 100% penetranci a léčba je indikována i u zcela asymptomatického nosiče mutace obou alel genu ATP7B. Léčbu je třeba zahájit co nejdříve, neboť s každým oddálením hrozí ireverzibilní poškození. Neléčená WN je fatální a pacienti umírají na jaterní selhání či komplikace imobilizace způsobené neurologickými projevy. Při včasně nasazené terapii je prognóza pacientů dobrá a délka života není oproti zdravé populaci zkrácena [6,46]. Nadále tedy platí, že nejčastější příčinou úmrtí u WN je pozdní diagnóza [47]. Zavedená léčba by měla probíhat celoživotně, a to i během těhotenství.
U symptomatických pacientů je v ČR lék první volby penicilamin. Zlepšení příznaků nelze po jeho nasazení očekávat dříve než po 3– 6 měsících a zlepšování trvá obvykle 1– 2 (maximálně tři) roky, poté se většinou neurologické příznaky ustálí. Posléze lze snížit dávku penicilaminu nebo přejít na léčbu zinkem. V iniciální fázi léčby cheláty dochází u 10– 50 % pacientů ke zhoršení neurologických příznaků. Většinou je přechodné, ale byly popsány i případy, u kterých se obtíže v dalším průběhu nezlepšily [48]. Dle některých souborů může docházet ke zhoršování neurologické symptomaticky i přes optimální léčbu až ve 24 % případů [15]. Dystonie a typická facies byly identifikovány jako příznaky nejvíce rezistentní k léčbě a jejich přítomnost je spojena s horší prognózou. Naopak forma s dominujícím tremorem je nejlépe ovlivnitelná terapií [16]. Ukazuje se, že průběh hepatální a neurologické symptomatiky v průběhu léčby spolu nekorelují, a je tedy možné, že patofyziologie poškození mozku a jater u WN jsou odlišné [12,15].
U presymptomatických pacientů se preferuje nasazení zinku, zejména u dětí mladších tří let. Někteří autoři preferují zinek i pro neuropsychiatrickou formu, zatímco pro hepatální formu dávají přednost penicilaminu [49]. Kontroverze panují ohledně současného podávání penicilaminu a zinku u neuropsychiatrické formy WN [26].
U pacientů s reziduálním neurologickým deficitem se uplatňuje i symptomatická léčba klinických obtíží pacienta [50]. V případě parkinsonského syndromu je na místě vyzkoušet dopaminergní terapii, levodopu či dopaminergního agonistu. Efekt může mít i amantadin. Fokální dystonii je obvykle možno příznivě ovlivnit botulotoxinem, generalizovanou dystonii anticholinergním přípravkem. Staticko‑kinetický tremor horních končetin může reagovat na primidon či clonazepam.
Problém je poměrně častá non‑compliance pacientů [51]. Pacienti by měli být upozorněni na riziko vysazení léčby, které spočívá v zhoršení neurologických i jaterních příznaků. Monitoring léčby je důležitý pro sledování ústupu klinických a laboratorních známek onemocnění, compliance pacientů a pro správné nastavení dávky farmak zajišťující optimální vylučování mědi z organizmu. Doporučený interval sledování je minimálně dvakrát ročně, vždy s vyšetřením krevního obrazu, chemickým vyšetřením moči, volné mědi v séru a vylučování mědi močí za 24 hod. Kupriuréza iniciálně po zavedení terapie penicilaminem výrazně stoupne, ale při chronické léčbě by se hodnoty měly pohybovat v rozmezí 3– 8 µmol/ 24 hod. Hodnoty pod 3 µmol/ 24 hod mohou znamenat buď non‑compliance nebo hypokupremii způsobenou léčbou. Zvýšená volná sérová měď v tomto případě svědčí pro non‑compliance. Příznaky hypokupremie jsou sideroblastická anémie, neutropenie, hyperferitinemie a myelopatie. Po dvou dnech vysazení penicilaminu či trientinu by při adekvátní léčbě by neměla kupriuréza překročit hodnotu 1,6 µmol/ l [13].
Při léčbě zinkem by kupriuréza neměla překročit hodnotu 1,6 µmol/ 24 hod a volná sérová měď by se měla pohybovat ve fyziologickém rozmezí. K potvrzení dobré compliance lze změřit odpad zinku močí za 24 hod.
Pacient by měl být pravidelně sledován také oftalmologem a vyšetřován pomocí MR mozku. Známkou non‑compliance je znovuobjevení se KF prstence, slunečnicové katarakty nebo zhoršení změn na MR mozku u pacienta, u něhož již tyto příznaky vymizely.
Závěr
Wilsonova nemoc je relativně vzácné, ale dobře léčitelné onemocnění s výbornou prognózou v případě časně zahájené terapie. Vzhledem k absenci doporučených postupů založených na randomizovaných studiích je vhodné, aby léčba a monitorování pacientů probíhaly ve specializovaných centrech se zkušenostmi s touto diagnózou. Projevy onemocnění mohou být velmi různorodé a je třeba na ně myslet u pacientů s jakýmkoliv extrapyramidovým či cerebelárním postižením, zejména v mladším věku.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Přijato k recenzi: 4. 6. 2013
Přijato do tisku: 9. 8. 2013
MUDr. Petr Dušek, Ph.D.
Neurologická klinika a Centrum klinických neurověd
1. LF UK a VFN
Kateřinská 30
120 00 Praha 2
e-mail: pdusek@gmail.com
Recenzenti:
doc. MUDr. David Školoudík, Ph.D.
MUDr. Martin Kuliha
MU Dr. Petr Dušek, Ph.D.

V letech 1996– 2003 absolovoval 3. LF UK v Praze a od té doby pracuje na Neurologické klinice 1. LF UK a VFN, postupně jako sekundární lékař, doktorand a odborný asistent téže kliniky. V roce 2009 získal atestaci z neurologie a v roce 2011 titul Ph.D. obhajobou dizertační práce na téma „Kortikální a subkortikální mechanizmy percepce času“. V témže roce absolvoval stáž v Parkinson’s Disease Center and Movement Disorders Clinic na Baylor College of Medicine v Houstonu, USA. Od roku 2013 pracuje na částečný úvazek jako výzkumný pracovník na klinice neuroradiologie v Göttingen a v Max- Delbrück centru v Berlíně. Je autorem nebo spoluautorem asi 25 článků v recenzovaných časopisech, několika kapitol v monografiích a na webových vzdělávacích portálech, dále je recenzentem řady zahraničních periodik. Jeho subspecializacemi v neurologii jsou extrapyramidová onemocnění a zobrazovací metody. V současnosti je jeho hlavním výzkumným zájmem zejména vztah mezi akumulací železa a těžkých kovů v CNS a rozvojem neurodegenerace. Je jedním ze zakládajících členů Neuropsychiatrického fóra, v rámci kterého pravidelně spolupořádá odborné konference zaměřené na edukaci a mezioborovou spolupráci v rámci neurovědních oborů.
Sources
1. Tanzi RE, Petrukhin K, Chernov I, Pellequer JL, Wasco W, Ross B et al. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat Genet 1993; 5(4): 344– 350.
2. Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW. The Wilson disease gene is a putative copper transporting P‑type ATPase similar to the Menkes gene. Nat Genet 1993; 5(4): 327– 337.
3. Coffey AJ, Durkie M, Hague S, McLay K, Emmerson J,Lo C et al. A genetic study of Wilson‘s disease in the United Kingdom. Brain 2013; 136(Pt 5): 1476– 1487.
4. Walshe JM. Wilson‘s disease. The presenting symptoms. Arch Dis Child 1962; 37: 253– 256.
5. Akil M, Schwartz JA, Dutchak D, Yuzbasiyan‑ Gurkan V, Brewer GJ. The psychiatric presentations of Wilson‘s disease. J Neuropsychiatry Clin Neurosci 1991; 3(4): 377– 382.
6. Bruha R, Marecek Z, Pospisilova L, Nevsimalova S, Vitek L, Martasek P et al. Long‑term follow‑up of Wilson disease: natural history, treatment, mutations analysis and phenotypic correlation. Liver Int 2011; 31(1): 83– 91.
7. Lalioti V, Sandoval I, Cassio D, Duclos‑ Vallee JC. Molecular pathology of Wilson‘s disease: a brief. J Hepatol 2010; 53(6): 1151– 1153.
8. Nagasaka H, Inoue I, Inui A, Komatsu H, Sogo T, Murayama K et al. Relationship between oxidative stress and antioxidant systems in the liver of patients with Wilson disease: hepatic manifestation in Wilson disease as a consequence of augmented oxidative stress. Pediatr Res 2006; 60(4): 472– 477.
9. Brewer GJ. A brand new mechanism for copper toxicity. J Hepatol 2007; 47(4): 621– 622.
10. Merle U, Stremmel W. Copper toxicity in Wilson disease explained in a new way. Hepatology 2011; 54(1): 358– 360.
11. Ferenci P, Czlonkowska A, Merle U, Ferenc S, Gromadzka G, Yurdaydin C et al. Late‑ onset Wilson‘s disease. Gastroenterology 2007; 132(4): 1294– 1298.
12. Lee BH, Kim JH, Lee SY, Jin HY, Kim KJ, Lee JJ et al. Distinct clinical courses according to presenting phenotypes and their correlations to ATP7B mutations in a large Wilson‘s disease cohort. Liver Int 2011; 31(6): 831– 839.
13. European Association for Study of Liver. EASL Clinical Practice Guidelines: Wilson‘s disease. J Hepatol 2012; 56(3): 671– 685.
14. Ala A, Walker AP, Ashkan K, Dooley JS, Schilsky ML. Wilson‘s disease. Lancet 2007; 369(9559): 397– 408.
15. Merle U, Schaefer M, Ferenci PStremmel W. Clinical presentation, diagnosis and long‑term outcome of Wilson‘s disease: a cohort study. Gut 2007; 56(1): 115– 120.
16. Burke JF, Dayalu P, Nan B, Askari F, Brewer GJ, Lorincz MT. Prognostic significance of neurologic examination findings in Wilson disease. Parkinsonism Relat Disord 2011; 17(7): 551– 556.
17. Barthel H, Hermann W, Kluge R, Hesse S, Collingridge DR, Wagner A et al. Concordant pre‑ and postsynaptic deficits of dopaminergic neurotransmission in neurologic Wilson disease. AJNR Am J Neuroradiol 2003; 24(2): 234– 238.
18. Lorincz MT. Neurologic Wilson‘s disease. Ann N Y Acad Sci 2010; 1184: 173– 187.
19. Akil M, Brewer GJ. Psychiatric and behavioral abnormalities in Wilson‘s disease. Adv Neurol 1995; 65: 171– 178.
20. Taly AB, Meenakshi‑ Sundaram S, Sinha S, Swamy HS, Arunodaya GR. Wilson disease: description of 282 patients evaluated over 3 decades. Medicine (Baltimore) 2007; 86(2): 112– 121.
21. Srinivas K, Sinha S, Taly AB, Prashanth LK, Arunodaya GR, Janardhana Reddy YC et al. Dominant psychiatric manifestations in Wilson‘s disease: a diagnostic and therapeutic challenge! J Neurol Sci 2008; 266(1– 2): 104– 108.
22. Svetel M, Potrebic A, Pekmezovic T, Tomic A, Kresojevic N, Jesic R et al. Neuropsychiatric aspects of treated Wilson‘s disease. Parkinsonism Relat Disord 2009; 15(10): 772– 775.
23. Eisenbach C, Sieg O, Stremmel W, Encke J, Merle U.Diagnostic criteria for acute liver failure due to Wilson disease. World J Gastroenterol 2007; 13(11): 1711– 1714.
24. Saito T. Presenting symptoms and natural history of Wilson disease. Eur J Pediatr 1987; 146(3): 261– 265.
25. Walshe JM. The eye in Wilson disease. QJM 2011; 104(5): 451– 453.
26. Roberts EA, Schilsky ML, American Association for Study of Liver D. Diagnosis and treatment of Wilson disease: an update. Hepatology 2008; 47(6): 2089– 2111.
27. Macintyre G, Gutfreund KS, Martin WR, Camicioli R,Cox DW. Value of an enzymatic assay for the determination of serum ceruloplasmin. J Lab Clin Med 2004; 144(6): 294– 301.
28. Martins da Costa C, Baldwin D, Portmann B, Lolin Y, Mowat AP, Mieli‑ Vergani G. Value of urinary copper excretion after penicillamine challenge in the diagnosis of Wilson‘s disease. Hepatology 1992; 15(4): 609– 615.
29. Ferenci P, Steindl‑ Munda P, Vogel W, Jessner W, Gschwantler M, Stauber R et al. Diagnostic value of quantitative hepatic copper determination in patients with Wilson‘s Disease. Clin Gastroenterol Hepatol 2005; 3(8): 811– 818.
30. Kenney SM, Cox DW. Sequence variation database for the Wilson disease copper transporter, ATP7B. Hum Mutat 2007; 28(12): 1171– 1177.
31. Vrabelova S, Letocha O, Borsky M, Kozak L. Mutation analysis of the ATP7B gene and genotype/ phenotype correlation in 227 patients with Wilson disease. Mol Genet Metab 2005; 86(1– 2): 277– 285.
32. Stapelbroek JM, Bollen CW, van Amstel JK, van Erpecum KJ, van Hattum J, van den Berg LH et al. The H1069Q mutation in ATP7B is associated with late and neurologic presentation in Wilson disease: results of a meta‑analysis. J Hepatol 2004; 41(5): 758– 763.
33. Czlonkowska A, Gromadzka G, Chabik G. Monozygotic female twins discordant for phenotype of Wilson‘s disease. Mov Disord 2009; 24(7): 1066– 1069.
34. Sinha S, Taly AB, Ravishankar S, Prashanth LK, Venugopal KS, Arunodaya GR et al. Wilson‘s disease: cranial MRI observations and clinical correlation. Neuroradiology 2006; 48(9): 613– 621.
35. Huang CC, Chu NS. Psychosis and epileptic seizures in Wilson‘s disease with predominantly white matter lesions in the frontal lobe. Parkinsonism Relat Disord 1995; 1(1): 53– 58.
36. Semnic R, Svetel M, Dragasevic N, Petrovic I, Kozic D,Marinkovic J et al. Magnetic resonance imaging morphometry of the midbrain in patients with Wilson disease. J Comput Assist Tomogr 2005; 29(6): 880– 883.
37. Prashanth LK, Sinha S, Taly ABVasudev MK. Do MRI features distinguish Wilson‘s disease from other early onset extrapyramidal disorders? An analysis of 100 cases. Mov Disord 2010; 25(6): 672– 678.
38. Kim TJ, Kim IO, Kim WS, Cheon JE, Moon SG, Kwon JW et al. MR imaging of the brain in Wilson disease of childhood: findings before and after treatment with clinical correlation. AJNR Am J Neuroradiol 2006; 27(6): 1373– 1378.
39. Huang CC, Chu NS. Resolution of cerebral white matter lesions following long‑term penicillamine therapy for Wilson‘s disease: report of a case. J Formos Med Assoc 1992; 91(6): 627– 629.
40. Walter U, Krolikowski K, Tarnacka B, Benecke R, Czlonkowska A, Dressler D. Sonographic detection of basal ganglia lesions in asymptomatic and symptomatic Wilson disease. Neurology 2005; 64(10): 1726– 1732.
41. Svetel M, Mijajlović M, Tomić A, Kresojević N, Pekmezović T, Kostić VS. Transcranial sonography in Wilson‘s disease. Parkinsonism Relat Disord 2012; 18(3): 234– 238.
42. Ferenci P, Caca K, Loudianos G, Mieli‑ Vergani G, Tanner S, Sternlieb I et al. Diagnosis and phenotypic classification of Wilson disease. Liver Int 2003; 23(3): 139– 142.
43. Weiss KH, Thurik F, Gotthardt DN, Schafer M, Teufel U, Wiegand F et al. Efficacy and Safety of Oral Chelators in Treatment of Patients with Wilson Disease. Clin Gastroenterol Hepatol 2013; 11(8): 1028– 1035.
44. Brewer GJ. Neurologically presenting Wilson‘s disease: epidemiology, pathophysiology and treatment. CNS Drugs 2005; 19(3): 185– 192.
45. Catana AM, Medici V. Liver transplantation for Wilson disease. World J Hepatol 2012; 4(1): 5– 10.
46. Stremmel W, Meyerrose KW, Niederau C, Hefter H,Kreuzpaintner G, Strohmeyer G. Wilson disease: clinical presentation, treatment, and survival. Ann Intern Med 1991; 115(9): 720– 726.
47. Walshe JM. Cause of death in Wilson disease. Mov Disord 2007; 22(15): 2216– 2220.
48. Brewer GJ, Terry CA, Aisen AM, Hill GM. Worsening of neurologic syndrome in patients with Wilson‘s disease with initial penicillamine therapy. Arch Neurol 1987; 44(5): 490– 493.
49. Wiggelinkhuizen M, Tilanus ME, Bollen CW, Houwen RH. Systematic review: clinical efficacy of chelator agents and zinc in the initial treatment of Wilson disease. Aliment Pharmacol Ther 2009; 29(9): 947– 958.
50. Holscher S, Leinweber B, Hefter H, Reuner U, Gunther P, Weiss KH et al. Evaluation of the symptomatic treatment of residual neurological symptoms in Wilson disease. Eur Neurol 2010; 64(2): 83– 87.
51. Walshe JM, Dixon AK. Dangers of non‑compliance in Wilson‘s disease. Lancet 1986; 1(8485): 845– 847.
52. Carta M, Mura G, Sorbello O, Farina G, Demelia L.Quality of Life and Psychiatric Symptoms in Wilson‘s Disease: the Relevance of Bipolar Disorders. Clin Pract Epidemiol Ment Health 2012; 8: 102– 109.
53. Starosta‑ Rubinstein S, Young AB, Kluin K, Hill G, Aisen AM, Gabrielsen T et al. Clinical assessment of 31 patients with Wilson‘s disease. Correlations with structural changes on magnetic resonance imaging. Arch Neurol 1987; 44(4): 365– 370.
54. Machado AA, Deguti MM, Genschel J, Cancado EL, Bochow B, Schmidt H et al. Neurological manifestations and ATP7B mutations in Wilson‘s disease. Parkinsonism Relat Disord 2008; 14(3): 246– 249.
55. Prashanth LK, Sinha S, Taly AB, Mahadevan A, Vasudev MK, Shankar SK. Spectrum of epilepsy in Wilson‘s disease with electroencephalographic, MR imaging and pathological correlates. J Neurol Sci 2010; 291(1– 2): 44– 51.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2013 Issue 5
Most read in this issue
- Wilson Disease
- Glioblastoma Multiforme – a Review of Pathogenesis, Biomarkers and Therapeutic Perspectives
- Tumefactive Variant of Multiple Sclerosis – Two Case Reports
- The 3F Test Dysarthric Profile – Normative Speach Values in Czech