
Dysembryoplastický neuroepiteliální tumor a jeho atypická varianta u dětí – kazuistiky
Dysembryoplastic Neuroepithelial Tumor and its Atypical Variant in Children – Case Reports
In this short summary, the authors describe an atypical form of dysembryoplastic neuroepithelial tumor (DNET) in children that has a different clinical, radiological and cytological profile that the typical DNET. Epileptic seizures with good response to radical surgical resection are the main clinical feature of a typical DNET. These tumors are typically in frontal or temporal locations. We describe three patients who underwent surgery during the last eight months, including a girl with an atypical form of DNET presenting with atypical clinical and, mainly, cytogenetic findings.
Key words:
neuroepithelial tumors – atypical forms
Authors:
D. Krahulík 1; M. Vaverka 1; L. Tučková 2; M. Köcher 3
Authors‘ workplace:
LF UP a FN Olomouc
Neurochirurgická klinika
1; LF UP a FN Olomouc
Ústav klinické a molekulární patologie
2; LF UP a FN Olomouc
Radiologická klinika
3
Published in:
Cesk Slov Neurol N 2013; 76/109(4): 492-496
Category:
Case Report
Overview
V následujícím krátkém sdělení autoři poukazují na atypickou formu dysembryoplastického neuroepiteliálního tumoru (DNET) u dětí s odlišným klinickým radiologickým, operačním a cytogenetickým profilem. Hlavním klinickým projevem DNET nádorů je epilepsie s dobrou odpovědí na resekční terapii. Tyto tumory rostou intrakortikálně s predominantní lokalizací v temporálním laloku. Autoři předkládají tři kazuistiky dětí s DNET tumorem operovaných během posledních osmi měsíců, přičemž jedna pacientka měla atypický klinický, pooperační a hlavně cytogenetický nález odpovídající v literatuře vzácně popisované atypické formě DNET.
Klíčová slova:
neuroepiteliální nádory – atypické formy
Úvod
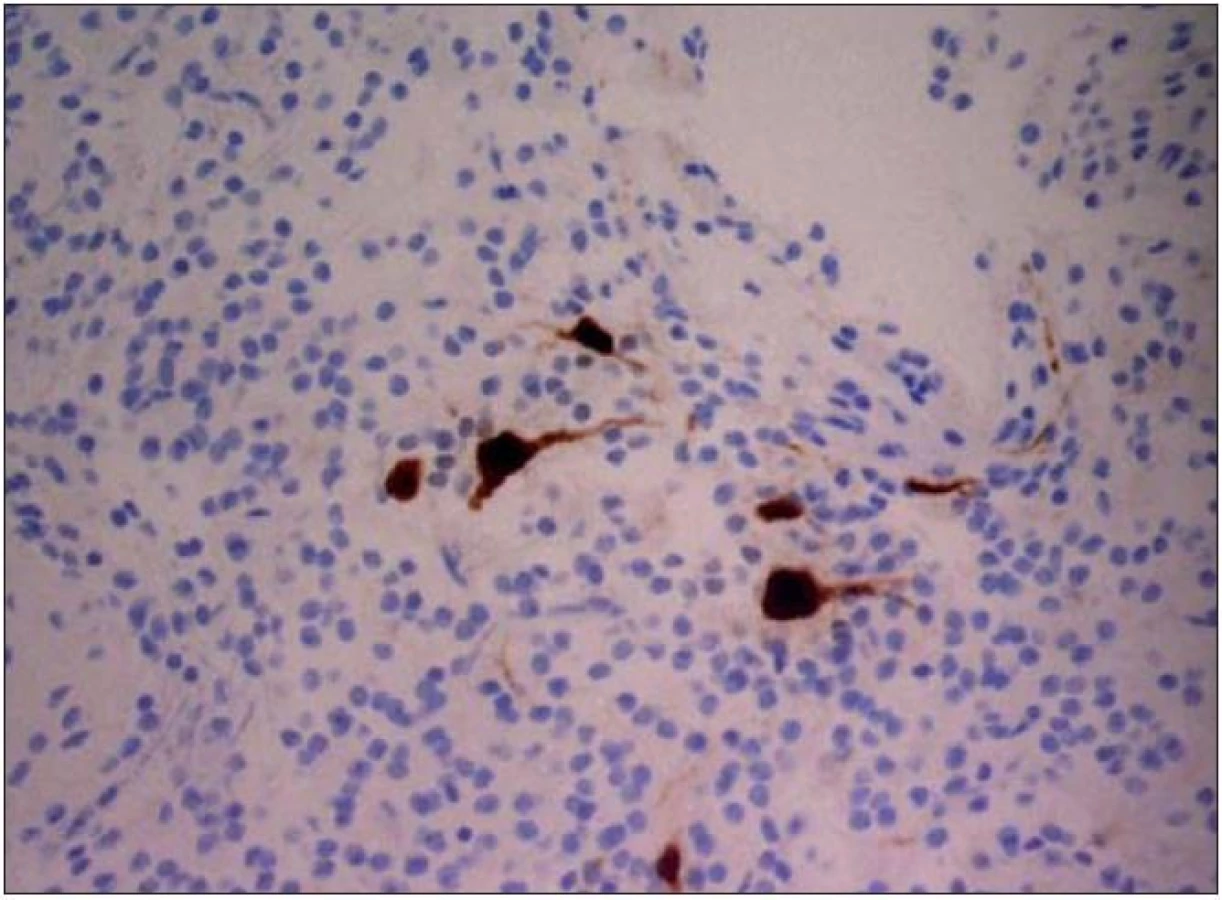
Dysembryoplastický neuroepiteliální tumor byl poprvé popsán v roce 1988 Daumas- Duportovou et al [1]. Tyto kortikálně uložené expanze se vyskytují nejčastěji u dětí a jejich první příznak bývá epileptický záchvat [2]. Popisované intraaxiální nádory jsou klasifikovány jako grade I dle WHO klasifikace a jejich struktura se skládá z oligodendrocytů navázaných na výběžky axonů plovoucí v mukoidní matrix (obr. 1, 2) [3]. Existují také odlišné typy DNET nádorů, které obsahují různě vyjádřené množství astrocytů a oligoastrocytů. Lokalizace DNET tumorů je ve většině případů supratentoriální s predominancí temporálního a frontálního laloku.

Základní zobrazovací metoda je magnetická rezonance, na které se zobrazují jako multilobulární masa často s cystickou složkou. DNET nádory jsou v T2 sekvencích hyperintenzivní a hypointenzivní v T1 vážených obrazech. Po podání kontrastní látky dochází k mírnému nebo častěji žádnému sycení tumoru. Ve FLAIR sekvenci mají tyto expanze smíšenou intenzitu [4]. U komplexních variant sledujeme kalcifikace [5].
Radikální neurochirurgický zákrok je hlavní terapeutická metoda bez nutnosti adjuvantní chemo- nebo radioterapie.
Kazuistiky
Tři dívky byly operovány během osmi měsíců na Neurochirurgické klinice LF UP a FN Olomouc. U všech dětí byl epileptický záchvat první příznak onemocnění a měly na magnetické rezonanci diagnostikovanou intraaxiální expanzi suspektní z DNET. Byla provedena radikální resekce ložisek s využitím elektromagnetické neuronavigace (Medtronic, USA) a histologicky byl potvrzen dysembryoplastický neuroepiteliální tumor. U všech nádorů bylo provedeno cytogenetické a molekulárněbiologické vyšetření. V třetí kazuistice byl prokázán atypický průběh onemocnění s odlišným cytogenetickým vyšetřením a klinickým průběhem, jak demonstrujeme na následujících kazuistických sděleních.
Kazuistika 1
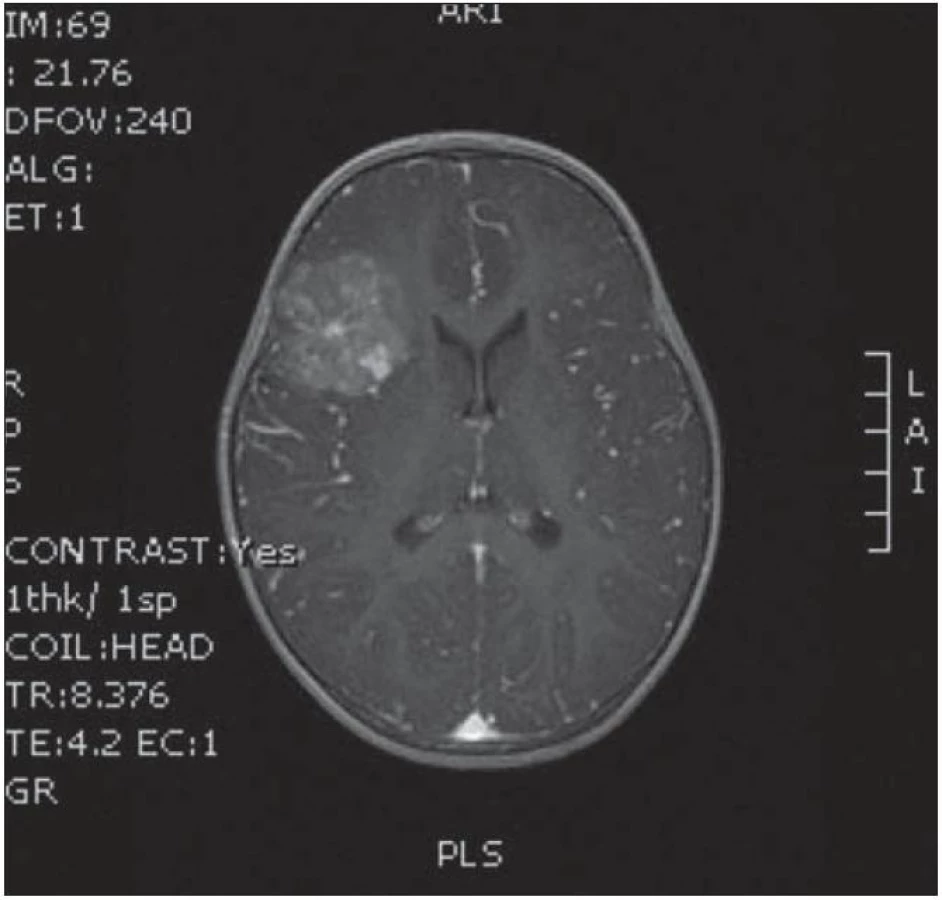
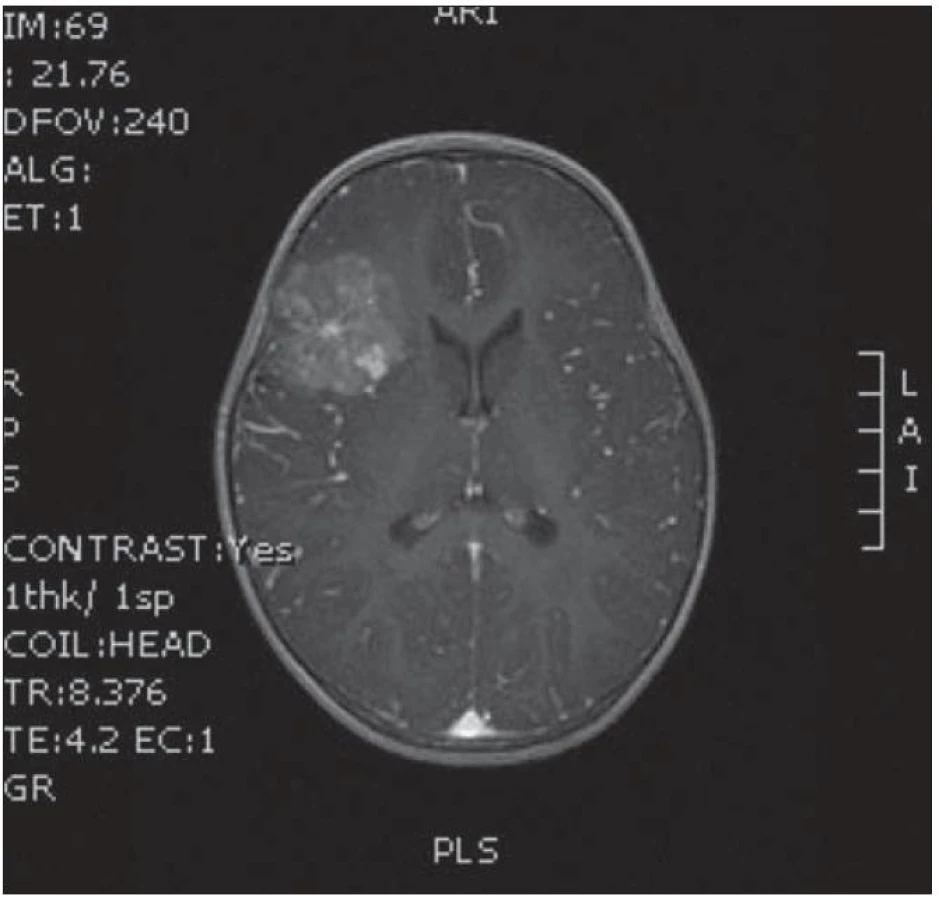
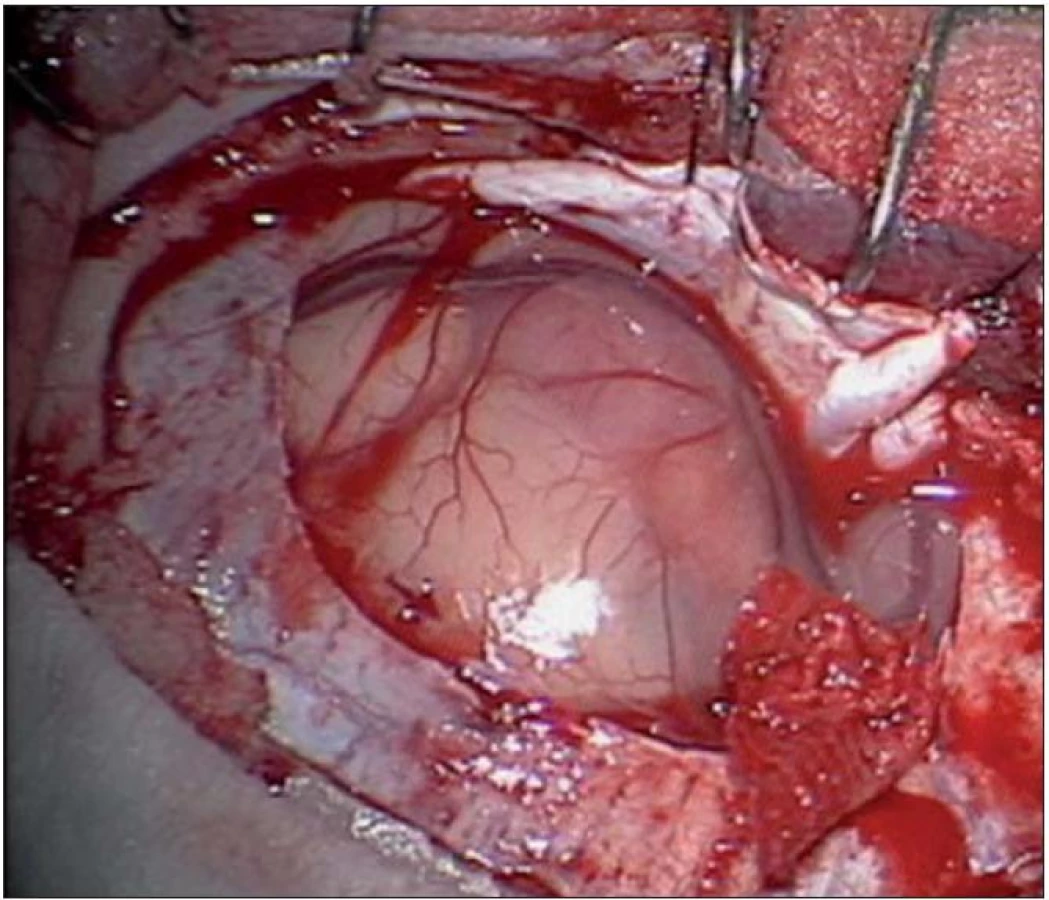
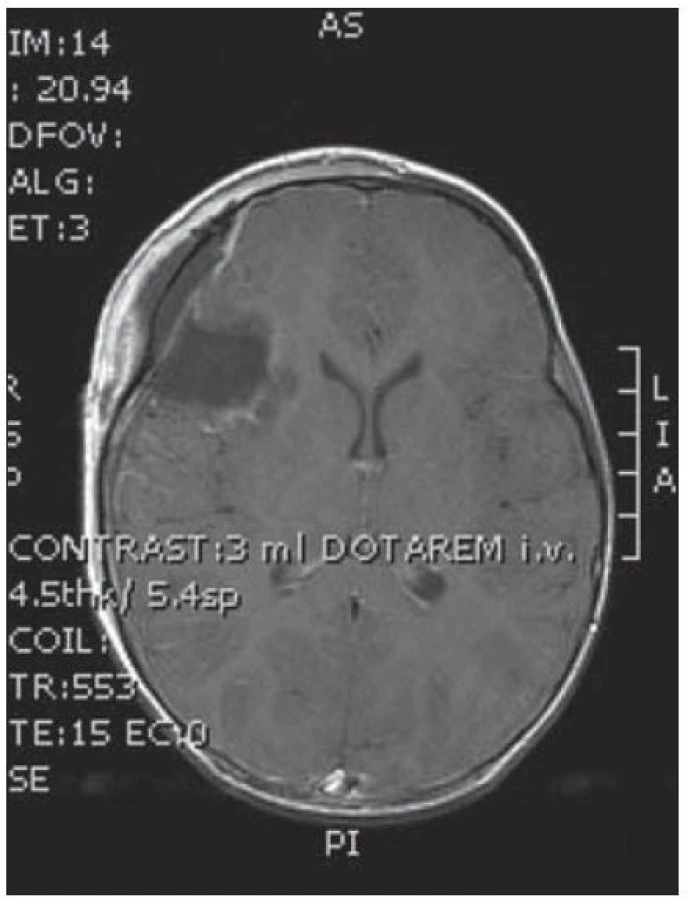
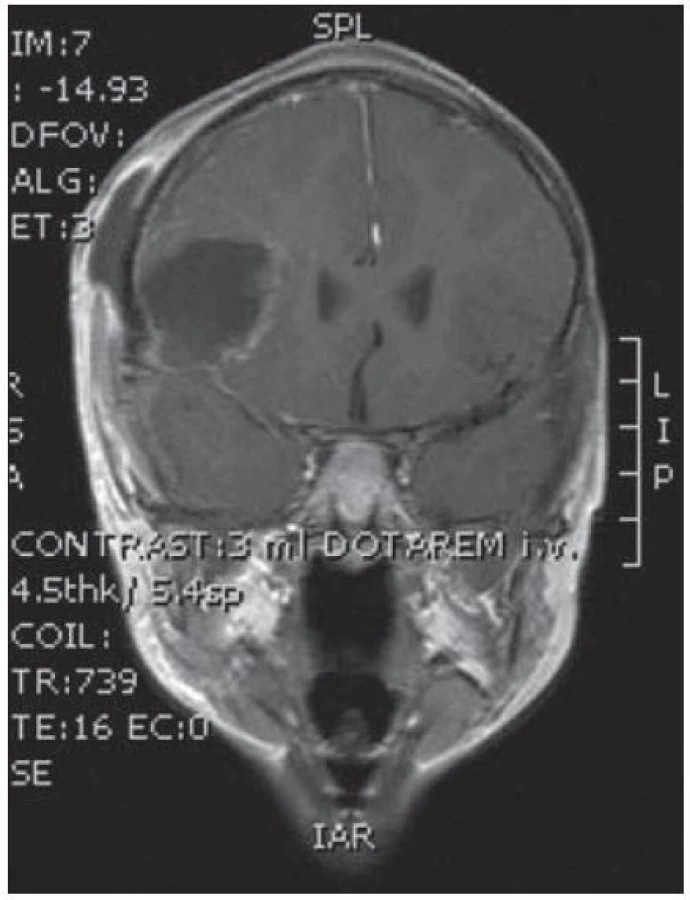
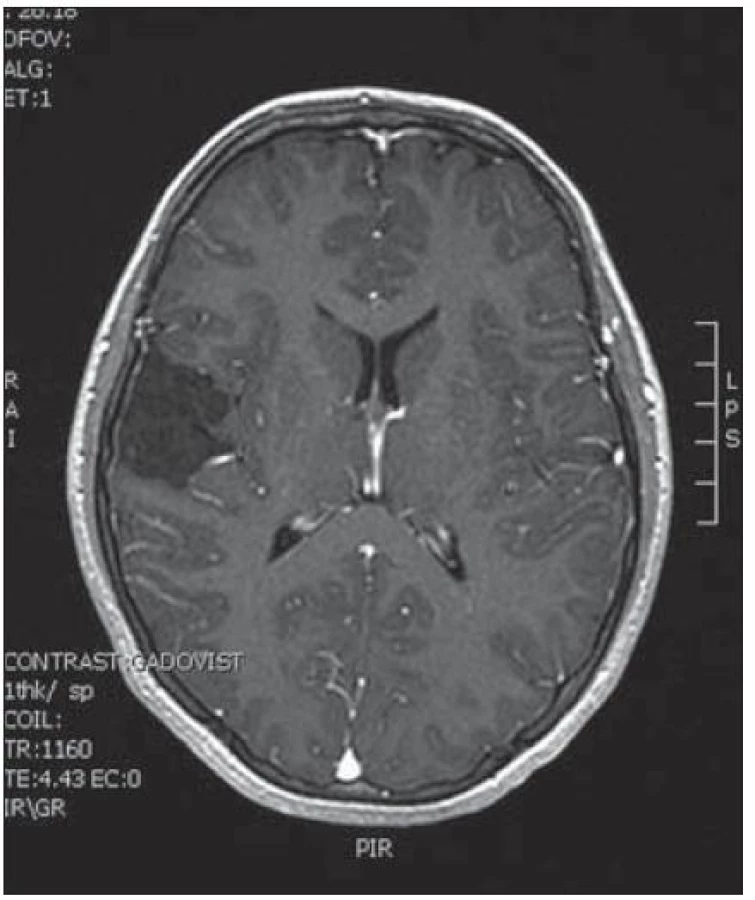
Dvouletá holčička byla léčena pro opakující se epiparoxyzmus s orofaciálními záškuby. Záchvaty ustaly po nasazení depakinu a doplněné MR vyšetření prokázalo heterogenní expanzi frontotemporálně vpravo (obr. 3, 4). Provedená radikální resekce měla nekomplikovaný průběh (obr. 5, 6). Pooperační MR kontrola dokumentovala radikální resekci bez rezidua tumoru (obr. 7, 8). Cytogenetické a histologické vyšetření identifikovalo tumor s oligodendrocytární složkou s místy nápadně spongioidně voštinovitou strukturou, kde v řídké prosáklé ALC (alcian) pozitivní hmotě nepravidelně rozhozeny neurocyty Synapt+ (synaptofyzin), NSE+ (neuron specifická enoláza) s pericelulárním halo bez satelitózy. Početně dominovaly oligodendrocyty a minoritně astrocyty, KI 67 negativní. Nádorová populace nevykazovala aberaci u žádného ze sledovaných markerů – status chromozomálních oblastí 9p21.3 (gen pro p16), 1p 36.3, 19 q 13, genu p53, RB1, n- MYC, BCR, chromozomu 10, 13,8, amplifikaci genu – MDM 2, EGFR 1, EGFR 19. Během sledování nedošlo k opakování epileptického paroxyzmu, EEG vyšetření bylo negativní a byla vysazena antiepileptická medikace.
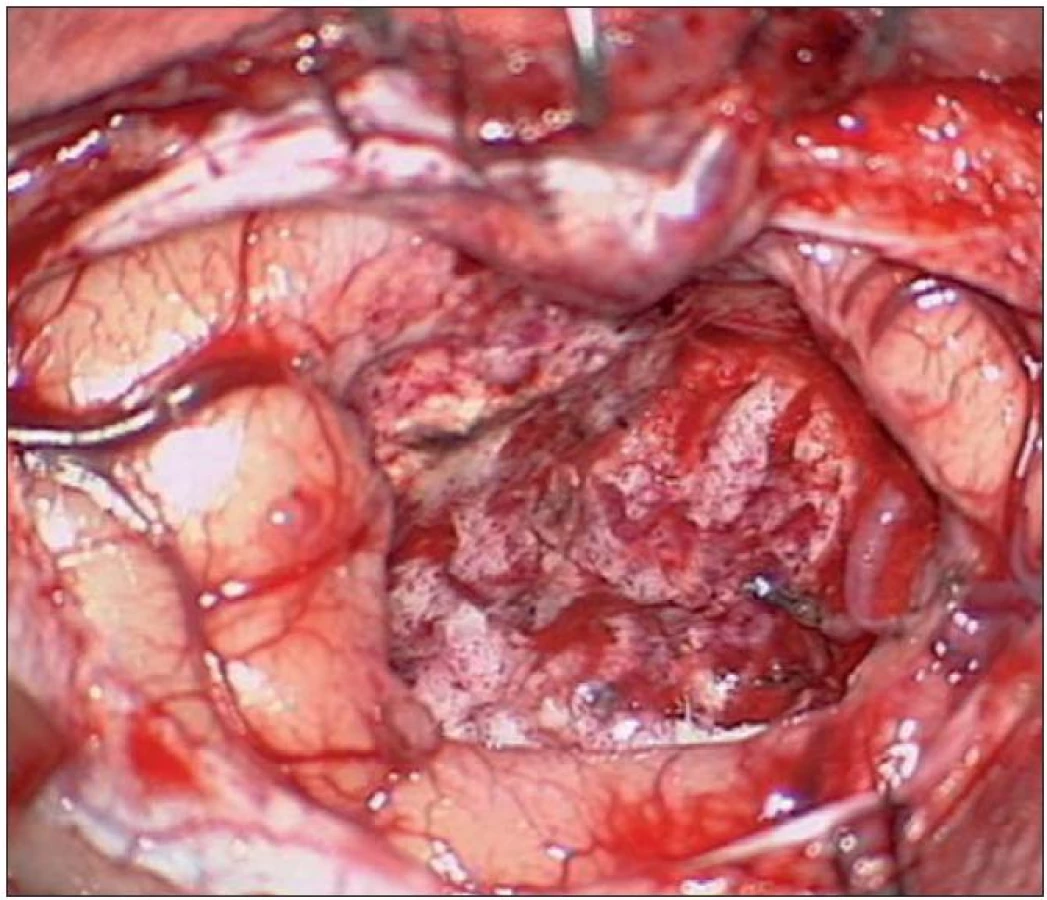
Kazuistika 2
Desetiletá dívka byla přijata na spádové neurologické oddělení pro deset minut trvající epileptický záchvat s tonicko‑klonickými záškuby levostranných končetin. V neurologickém nálezu byla frustní levostranná hemiparéza, oční pozadí bylo negativní a EEG ukázalo seskupení atypických vln v oblasti temporální vpravo. Doplněná magnetická rezonance zobrazila nesytící se ložisko spánkového laloku vpravo (obr. 9). Operační zákrok s využitím elektromagnetické navigace proběhl bez komplikací a pooperační MR bylo příznivé (obr. 10). Histologické vyšetření ukázalo struktury glioneurálního nádoru, v němž gliální složka oligodendrocytoidního vzhledu, místy naznačující nepravidelnou palisádaci, objemově převažovala nad složkou neuronální, jejíž gangliocyty, řídce dispergované v glii, byly kromě velikostní různorodosti jinak cytologicky nenápadné. Mnohotně jsou v nádoru přítomny kalkosférity různých velikostí, lineární kalcifikáty a kalcifikace ve stěnách cév. Imunofenotyp GFAP+ (gliální fibrilární kyselý protein) NFP– Synapt – NSE negativní. Při komplexním cytogenetickém a molekulárněbiologickém vyšetření nádorová populace nevykazovala aberaci u žádného ze sledovaných markerů (status chromozomálních oblastí 9p21.3/ gen pro p16/ ), 1p 36.3, 19 q 13, genu p53, RB1, n-MYC, BCR, chromozomu 10, 13,8, amplifikaci genu – MDM 2, EGFR 1, EGFR 19. U pacientky nedošlo k opakování epileptického paroxyzmu a kontrolní EEG vyšetření neprokázalo patologickou aktivitu.
Kazuistika 3
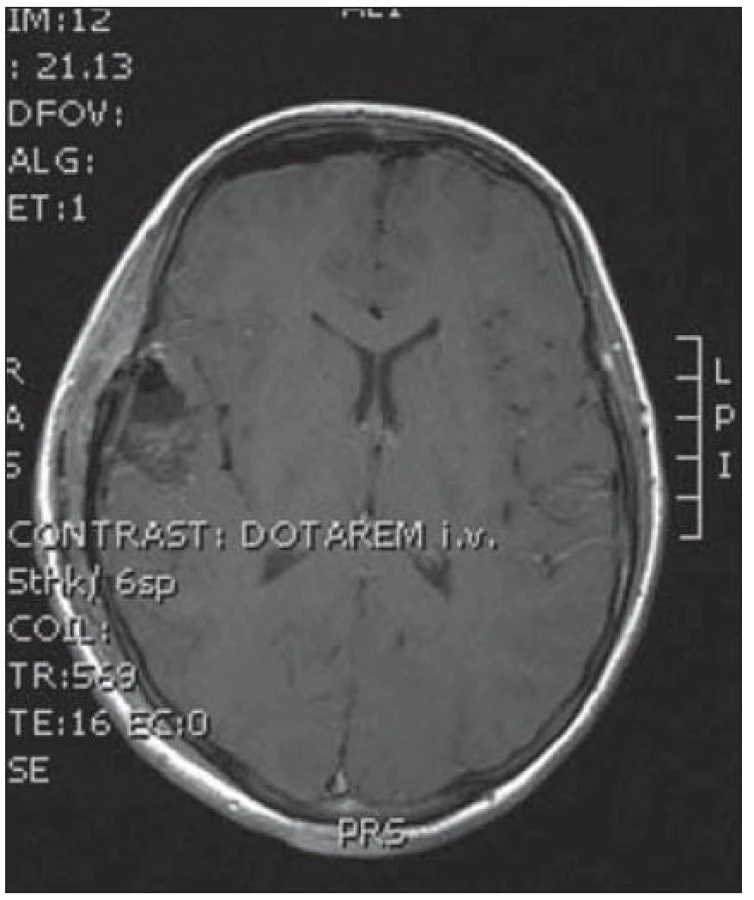
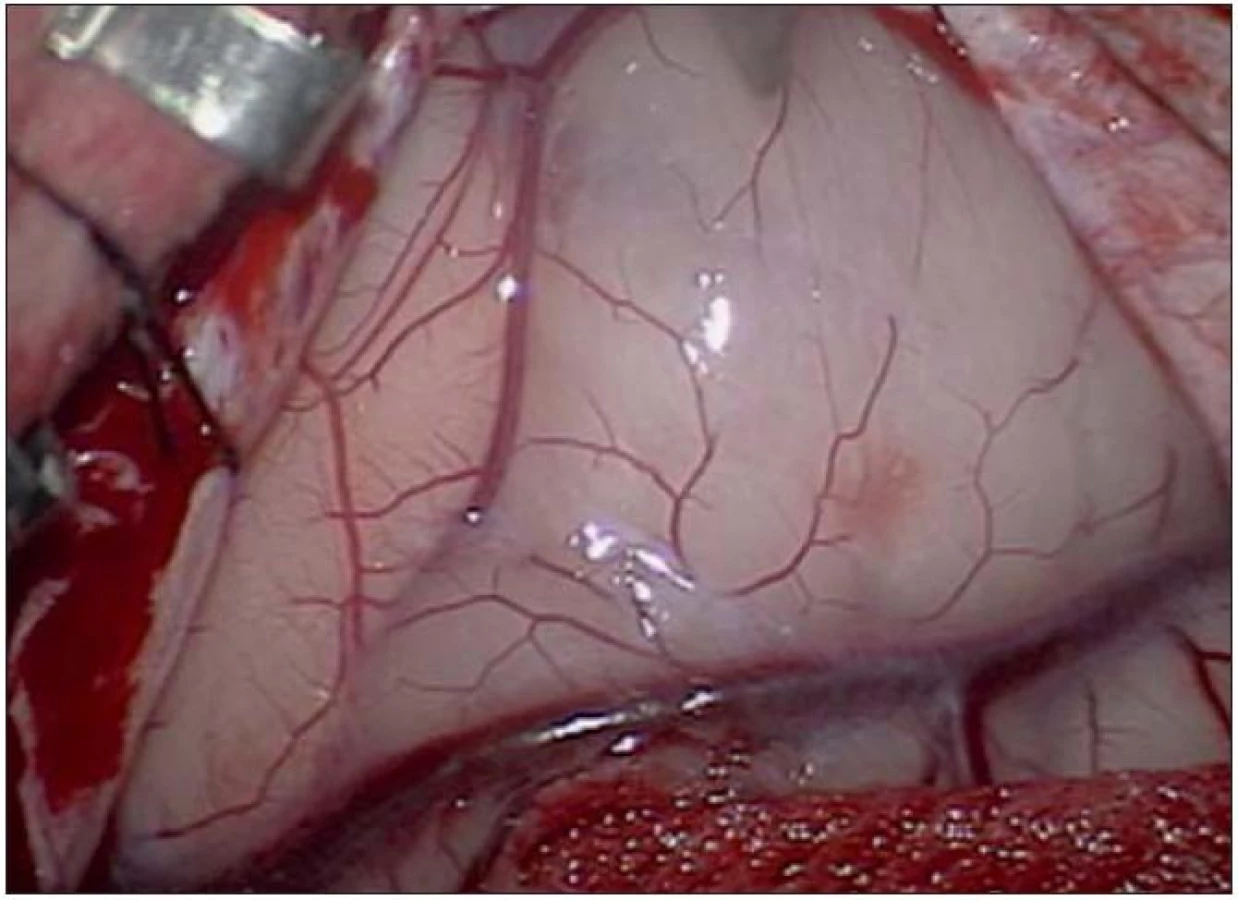
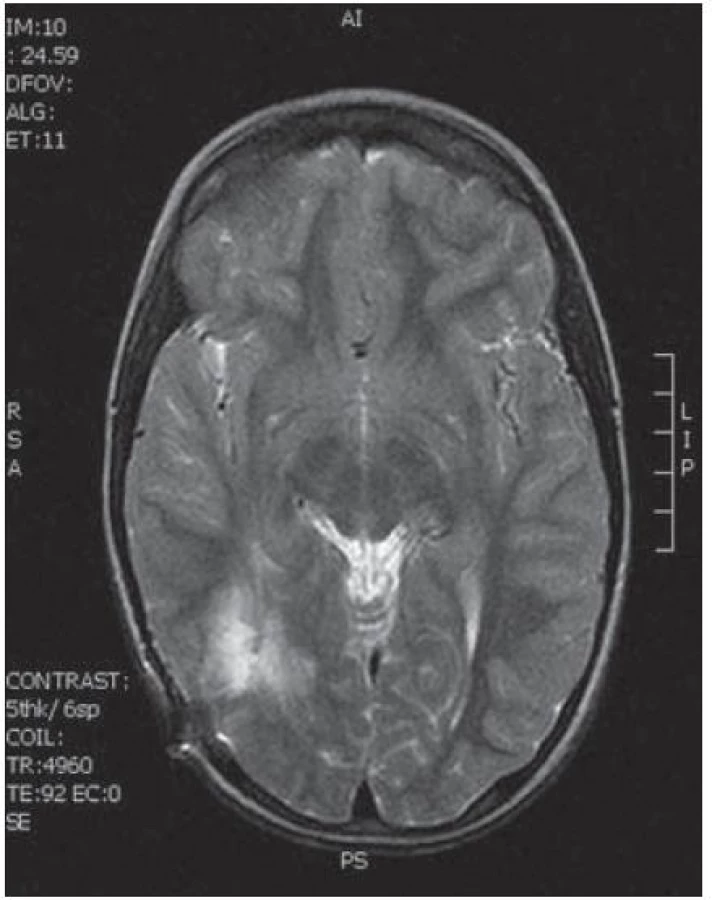
Poslední pacientka s atypickým průběhem byla sedmiletá dívka s měsíc trvajícími stavy areaktivity. EEG vyšetření mělo asymetrii pozadí mezi hemisférami, již od počátku patrné nepravidelné ostré vlny s maximem fronto- temporo‑parietálně vpravo – jednalo se o PSW (Peek Strong Wave) vysoké amplitudy s tendencí generalizovat. Nasazena antiepileptická léčba timonilem a magnetická rezonance odhalila expanzi temporo‑parietálně vpravo se sycením po k. l. (obr. 11, 12). Dívka byla operována, bez použití EECoG, s dosažením kompletní resekce tumoru, jak ukazuje pooperační MR T2 zobrazení tři měsíce po výkonu (obr. 13– 14). Pooperační průběh byl komplikován opakovaným epiparoxyzmem a bylo nutné upravit antiepileptickou léčbu na dvojkombinaci antiepileptik. Vyšetření perimetru bylo bez výpadku a ostatní neurologický nález v normě. Histologické vyšetření mělo hojnou oligodendrocytární složku s místy nápadně spongioidně voštinovitou strukturou, kde v řídké prosáklé ALC pozitivní hmotě nepravidelně rozhozeny neurocyty (Synapt+, NSE+) s pericelulárním halo bez satelitózy. Početně byly dominující oligodendrocyty a minoritní astrocyty, KI 67negativní. V cytogenetickém vyšetření nádorová populace vykazovala odlišnosti od předchozích pacientek, a to deleci na krátkém rameni chromozomu 9 (9p21.3, gen pro onkosupresor p16 a p15), monozomii chromozomu 10, deleci tumor supresorového genu p53 a genu RB1 jako důsledek monosomie chromozomu 13. Kontrolní EEG vyšetření mělo zmírněnou, ale stále přetrvávající lehkou aktivitu v resekované oblasti. V půlročním sledování se epileptický záchvat opakoval pouze jednou během prvních dvou měsíců a poté již ne.


Diskuze
DNET nádory se predominantně vyskytují v temporálním a frontálním laloku a jsou obvykle dobře ohraničené, kortikálně uložené, s velkou variabilitou velikosti a tvaru [6]. Diferenciálně diagnosticky dle MR nálezu musíme zvažovat gangliocytom, pilocytický astrocytom, pleomorfní xantoastrocytom či angiocentrický gliom [7].Cystické složky u gangliocytomu a pilocytického astrocytomu bývají více nepravidelného tvaru a distribuce, než obvykle sledujeme u DNET. Kontrastní sycení u DNET expanzí je nejčastěji nodulární, prstencovité či heterogenní na rozdíl od pleomorfního astrocytomu, kde sledujeme povrchový meningocerebrální enhancement [5]. U našich pacientů jsme se rozhodli přímo k radikálnímu operačnímu zákroku bez histologické verifikace biopsií, přestože někteří autoři ji doporučují provádět k získání histologické diagnózy [8]. Parmar et al [9] ve své práci popisují radiologickou charakteristiku DNET tumorů ve FLAIR zobrazení, která by mohla být pro tyto léze charakteristická. Tyto nálezy bývají popisovány jako gyriformní expanzivita a vysoce signální okraj expanze. Průměrný počáteční věk klinické prezentace u DNET nádorů je devět let s literaturně uváděnou lehce mužskou predominancí [1], kterou jsme ale v naší minisérii nezaznamenali. První příznak je nejčastěji fokální epileptický záchvat, ale byly popsány i případy klinické prezentace s Lennox- Gastautovým syndromem nebo pouze cefaleou [10,11]. Výsledky po radikálním neurochirurgickém zákroku jsou velice dobré a histologické a cytologické vyšetření pro další sledování je velice důležité [11– 13]. U typických DNET nádorů prezentovaných v prvních dvou případech a v souladu s literaturou dojde po exstirpaci k normalizaci EEG nálezu a je možné vysazení antiepileptické medikace. Pacienti jsou sledováni prostřednictvím MR vyšetření v intervalu 3, 6 a 12 měsíců. U poslední dívky byla epilepsie refrakterní a po operační terapii byl EEG nález stále patologický. I přes terapii se opakovaly záchvaty a bylo nutné navýšení a dvojkombinace antiepileptických preparátů. Cytogenetické, molekulárněbiologické a histologické vyšetření prokázaly odlišné nálezy od předchozích případů shodných s atypickou formou DNET prezentovaných v literatuře [8], které sice vzácně, ale přesto mohou mít maligní transformaci [14], a proto u tohoto typu nádoru doporučujeme častější kontroly pomocí magnetické rezonance každé tři měsíce během prvního roku a poté co půl roku.
Závěr
Výše popisované kazuistiky měly podobný předoperační klinický průběh, MR zobrazení, perioperační nález a histologické vyšetření. Refrakternost epilepsie a její pooperační recidivu u poslední kazuistiky vysvětlilo cytologické vyšetření se zcela odlišným nálezem oproti předchozím dvěma pacientkám. Tento nález odpovídá v literatuře popisované vzácné atypické formě DNET nádoru. Je velice důležité kompletní cytogenetické a molekulárněbiologické vyšetření, které nám tuto formu dokáže odlišit, a zaměřit se na častější sledování a agresivnější biologickou povahu tohoto typu nádoru.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr. David Krahulík, Ph.D.
Neurochirurgická klinika
LF UP a FN
I. P. Pavlova 6
775 20 Olomouc
e-mail: david.krahulik@fnol.cz
Přijato k recenzi: 16. 8. 2012
Přijato do tisku: 6. 11. 2012
Sources
1. Daumas-Duport C, Scheithauer BW, Chodkiewicz JP, Laws ER jr, Vedrenne C. Dysembryoplastic neuroepithelial tumor: a surgically curable tumor of young patients with intractable partial seizures. Report of thirty- nine cases. Neurosurgery 1988; 23(5): 545– 556.
2. Kaye AH, Laws E jr (eds). Brain tumors: an encyclopedic approach. 2nd ed. New York: Churchill Livingstone 2001.
3. Campos AR, Clusmann H, von Lehe M, Niehusmann P, Becker AJ, Schramm J et al. Simple and complex dysembryoplastic neuroepithelial tumors (DNT) variants: clinical profile, MRI, and histopathology. Neuroradiology 2009; 51(7): 433– 443.
4. Ostertun B, Wolf HK, Campos MG, Matus C, Solymosi L, Elger CE et al. Dysembryoplastic neuroepithelial tumors: MR and CT evaluation. AJNR Am J Neuroradiol 1996; 17(3): 419– 430.
5. Urbach H. MRI of long‑term epilepsy‑associated tumors. Semin Ultrasound CT MR 2008; 29(1): 40– 46.
6. Raymond AA, Halpin SF, Alsanjari N, Cook MJ, Kitchen ND, Fish DR et al. Dysembryoplastic neuroepithelial tumor. Features in 16 patients. Brain 1994; 117(3): 461– 475.
7. Hamada H, Kurimoto M, Nagai S, Asahi T, HirashimaY, Endo S. A rare case of dysembryoplastic neuroepithelial tumour in occipital lobe presenting with only headache. J Clin Neurosci 2003; 10(2): 276– 278.
8. Bird- Lieberman G, Sethi K, Childs AM, Chumas P, Crimmins D, Ismail A et al. Diffuse hemispheric dysembryoplastic neuroepithelial tumor: a new radiological variant associated with early- onset severe epilepsy. J Neurosurg Pediatr 2011; 7(4): 416– 420.
9. Parmar HA, Hawkins C, Ozelame R, Chuang S, Rutka J,Blaser S. Fluid- attenuated inversion recovery ring sign as a marker of dyembryoplastic neuroepithelial tumors. J Comput Assist Tomogr 2007; 31(3): 348– 353.
10. Hamada H, Kurimoto M, Nagai S, Asahi T, Hirashima Y, Endo S. A rare case of dysembryoplastic neuroepithelial tumour in occipital lobe presenting with only headache. J Clin Neurosci 2003; 10(2): 276– 278.
11. Quarato PP, Gennaro GD, Manfredi M, Esposito V.Atypical Lennox- Gastaut syndrome successfully treated with removal of a parietal dysembryoplastic tumour. Seizure 2002; 11(5): 325– 329.
12. Drake J, Hoffman HJ, Kobayashi J, Hwang P, Becker LE. Surgical management of children with temporal lobe epilepsy and mass lesions. Neurosurgery 1987; 21(6): 792– 797.
13. Nolan MA, Sakuta R, Chuang N, Otsubo H, Rutka JT,Snead OC 3rd et al. Dysembryoplastic neuroepithelial tumors in childhood: long‑term outcome and prognostic features. Neurology 2004; 62(12): 2270– 2276.
14. Luyken C, Blümcke I, Fimmers R, Urbach H, Elger CE,Wiestler OD et al. The spectrum of long‑term epilepsy‑associated tumors: long‑term seizure and tumor outcome and neurosurgical aspects. Epilepsia 2003; 44(6): 822– 830.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2013 Issue 4
Most read in this issue
- Úskalí diagnostiky progresivní multifokální leukoencefalopatie u pacientů infikovaných lidským virem imunodeficience – kazuistiky
- Chirurgie baze lební (uvnitř minimonografie video)
- Neuropsychiatrický pohľad na Huntingtonovu chorobu
- Dysembryoplastický neuroepiteliální tumor a jeho atypická varianta u dětí – kazuistiky