
Parkinsonské fenotypy – na cestě k nové nozologii atypických parkinsonských syndromů
Parkinsonian Phenotypes – towards New Nosology of Atypical Parkinsonian Syndromes
Parkinson’s disease is still viewed as a nosological entity (G20), yet it covers a wide range of clinical phenotypes characterised by the presence of parkinsonian syndrome with variable expression of a range of symptoms – motor, cognitive and behavioural – and an uncertain course. It appears that, largely thanks to progress in the recognition of phenotype variability and its clinico-pathological correlations at an ultrastructural level, it will divide into several close but separate nosological entities. In the last 50 years certain atypical parkinsonian syndromes have already split off one by one – Parkinson’s disease dementia (PDD), progressive supranuclear palsy (PSP), multiple system atrophy (MSA), corticobasal degeneration (CBGD), diffuse Lewy body disease (DLBD), frontotemporal dementia (FTLD) and the endemic parkinsonian syndromes such as Guam and Guadeloupean parkinsonism. However, the atypical parkinsonian syndromes are still diagnosed on the basis of (mainly obsolete) clinical diagnostic criteria which, at the time of their creation, only partially reflected the real pathological nature of the disorder. Their replacement with newly-developed criteria created on the basis of clinico-pathological correlation should be given priority in the field of translational research in the immediate future.
Key words:
Parkinson’s disease – atypical parkinsonian syndromes, clinico-pathological correlation
Authors:
K. Farníková 1; M. Bareš 2; I. Nestrašil 3; P. Kaňovský 1
Authors‘ workplace:
Centrum pro diagnostiku a léčbu neurodegenerativních onemocnění, Neurologická klinika LF UP a FN Olomouc
1; Centrum pro abnormní pohyby a parkinsonizmus, 1. neurologická klinika LF MU a FN u sv. Anny v Brně
2; Department of Neurology, University of Minnesota, Minneapolis, USA
3
Published in:
Cesk Slov Neurol N 2011; 74/107(6): 641-653
Category:
Review Article
Overview
Parkinsonova nemoc je v současnosti stále ještě nozologickou jednotkou (G20), která však skrývá pestrou škálu fenotypů charakterizovaných přítomností parkinsonského syndromu s měnlivou akcentací jednotlivých symptomů, motorických, kognitivních i behaviorálních, a s velmi variabilním průběhem. Zdá se, že se tato jednotka do budoucna (díky dalšímu pokroku v poznání variability fenotypů a jejich klinickopatologických korelací na ultrastrukturální úrovni) bude rozpadat na několik podrobněji definovaných nozologických jednotek. Podobně se během posledních 50 let postupně diferencovaly atypické parkinsonské syndromy jako Parkinsonova nemoc s demencí (PDD), progresivní supranukleární paralýza (PSP), multisystémová atrofie (MSA), kortikobazální degenerace (CBGD), demence s Lewyho tělísky (DLBD), frontotemporální demence (FTD) a endemické atypické parkinsonské syndromy, jako je guamský parkinsonský komplex a guadeloupský parkinsonizmus. Atypické parkinsonské syndromy jsou však stále ještě diagnostikovány na základě klinických diagnostických kritérií, která v době svého vzniku jen částečně reflektovala pravou patologickou podstatu poruchy; jejich nahrazení kritérii vzniklými na základě klinickopatologických korelací by mělo být hlavním úkolem výzkumu v této oblasti v příštích letech.
Klíčová slova:
Parkinsonova nemoc – atypické parkinsonské syndromy, klinickopatologická korelace
Úvod
Parkinsonův popis nemoci, která již více než sto let nese jeho jméno, zahrnuje dodnes udávanou triádu příznaků: klidový třes, ztuhlost (rigiditu) a povšechné zpomalení pohybu (bradykinezi), která byla až v posledních letech doplněna na tetrádu příznakem posturální instability [1,2]. Parkinsonova nemoc, dříve pokládaná za prototyp parkinsonského „hypokineticko-rigidního“ syndromu, byla v téže době považována za klinický korelát tzv. status lacunaris cerebri, kdy je oblast bazálních ganglií postižena drobnými mozkovými infarkty a mozková tkáň zde makroskopicky připomíná sýr s malými oky. Bohužel u určité části neurologické komunity zůstala tato představa parkinsonizmu a jeho příčin platná dodnes. Parkinsonova nemoc je nyní jak v evropské mezinárodní klasifikaci nemocí (MKN-10), tak i americkém Diagnostic and Statistic Manual (DSM-IV) klasifikována jako neurodegenerativní onemocnění, jehož klinické příznaky jsou důsledkem neurodegenerace více částí centrální nervové soustavy, nejvýznamněji patrně oblasti pars compacta substantiae nigrae. Již téměř deset let je však více či méně jasné, že klinický průběh Parkinsonovy nemoci je tak mnohotvárný, že vede k úvahám o rozpadu této klinické jednotky na další fenotypy [3,4]. Genotypicky se nozologická jednotka G20 rozpadá již od roku 1997, kdy byl popsán lokus PARK1, kódující manifestaci familiární alfa-1-synukleinopatie. Od té doby výzkum pokročil, takže takových lokusů je dnes známo 15 [5,6].
Nicméně dělení parkinsonského syndromu na fenotypy, které jsou od původního „hypokineticko-rigidního“ syndromu zásadně odlišné i patologicky, začalo už před téměř 50 lety.
Progresivní supranukleární paralýza (PSP, Steele-Richardson-Olszewski nemoc)
Začátkem 60. let minulého století si několik bystrých kanadských neurologů začalo všímat některých specifik klinického obrazu u pacientů, kteří byli léčeni pod diagnózou Parkinsonova nemoc. Patrně první z nich byl John Richardson, jenž u svých pacientů pozoroval „zvláštní“ typ pohledové obrny, dysexekutivní syndrom, progredující demenci, časté pády a absenci třesu. Jako spíše praktický – byť univerzitní – neurolog, který (asi) neměl čas na další metody paraklinického testování, přizval ke konzultaci dalšího kolegu, Johna Steelea. Tak došlo k tomu, že první z tzv. parkinson-plus syndromů se dnes kromě popisného názvu a zkratky (Progressive Supranuclear Palsy, PSP) nazývá nemocí Steele-Richardson-Olszewski [7,8]. Jerzy Olszewski byl patolog, který poprvé popsal dnes již charakteristický obraz PSP; tehdy pochopitelně bez zmínky o patologické agregaci tau proteinů. Je však otázka, které z podob nemoci toto jméno náleží, neboť následující vývoj poznání přinesl (před asi šesti lety) další rozdělení nozologické jednotky PSP na tzv. PSP Richardsonova typu a PSP parkinsonského typu, a o dva roky později byl přidán další fenotyp, tzv. pure akinesia with gait freezing [9]. Musíme zde však uvést, že John Steele na posledním světovém neurologickém kongresu sám navrhl, aby se porucha nadále klasifikovala jako jedna nozologická jednotka a aby se jmenovala Richardsonova nemoc (obr. 1) [10].
![Příloha 1. Klinická diagnostická kritéria progresivní supranukleární paralýzy [68].](https://pl-master.mdcdn.cz/media/image/6c07a6319d2ae23cf631ddf68c3975ba.jpeg?version=1537790321)

Multisystémová atrofie (MSA)
Další v řadě bylo onemocnění, které kombinovalo příznaky parkinsonizmu s autonomní dysfunkcí a mozečkovým a pyramidovým postižením. Porucha s dominantní autonomní symptomatikou byla již od poloviny 50. let nazývána Shy-Dragerovým syndromem, aniž kdy kdo podrobněji zkoumal bližší charakteristiky a kombinaci parkinsonských symptomů v rámci tohoto onemocněním [11]. Až v roce 1989 Nial Quinn dal této poruše název multisystémová atrofie a postuloval klinická kritéria tohoto onemocnění [12]. Pokrok však i v této době šel pomalu: v Čechách byla autoritami existence této nemoci zpochybňována ještě i po jejím prvním českém popisu v roce 1996 [13]. Za zhruba 20 let, která uplynula od prvního kompletního popisu onemocnění, diferenciace pokračovala. Quinnova původní kritéria jsou považována za obsolentní a nemoc je nyní popisována ve dvou variantách, tzv. MSA-C („cerebellar“) a MSA-P („parkinsonian“). Toto spíše arbitrární rozdělení je reflexí poměrně velkého počtu klinickopatologicky korelovaných případů, kdy typická patologie, totiž argyrofilní oligodendrogliální cytoplazmatické inkluze, byly nalezeny u případů s výrazně odlišným klinickým fenotypem (obr. 3) [14]. Rozdělením na fenotypy MSA-C a MSA-P jako bychom se obloukem vraceli do minulosti: MSA-C svými klinickými projevy a dalšími charakteristikami odpovídá zhruba tomu, co bylo v minulosti označováno jako „olivopontocerebellární atrofie“, ať již typu Pierre-Marie-Foix-Alajouanine, Stewart-Holmes či jakékoliv jiné kombinace autorských jmen. MSA-P potom odpovídá spíše tomu, čemu se říkalo „striatonigral degeneration“, byť toto označení nebylo používáno déle než několik let [15]. Je potřeba zde zdůraznit, že i dnešní kritéria předpokládají prakticky intaktní kognitivní funkce jak u formy MSA-C, tak i u formy MSA-P.
![Příloha 2. Klinická diagnostická kritéria multisystémové atrofie [69].](https://pl-master.mdcdn.cz/media/image/4a14c7268bdc556255cb73676c567b2e.jpeg?version=1537790321)
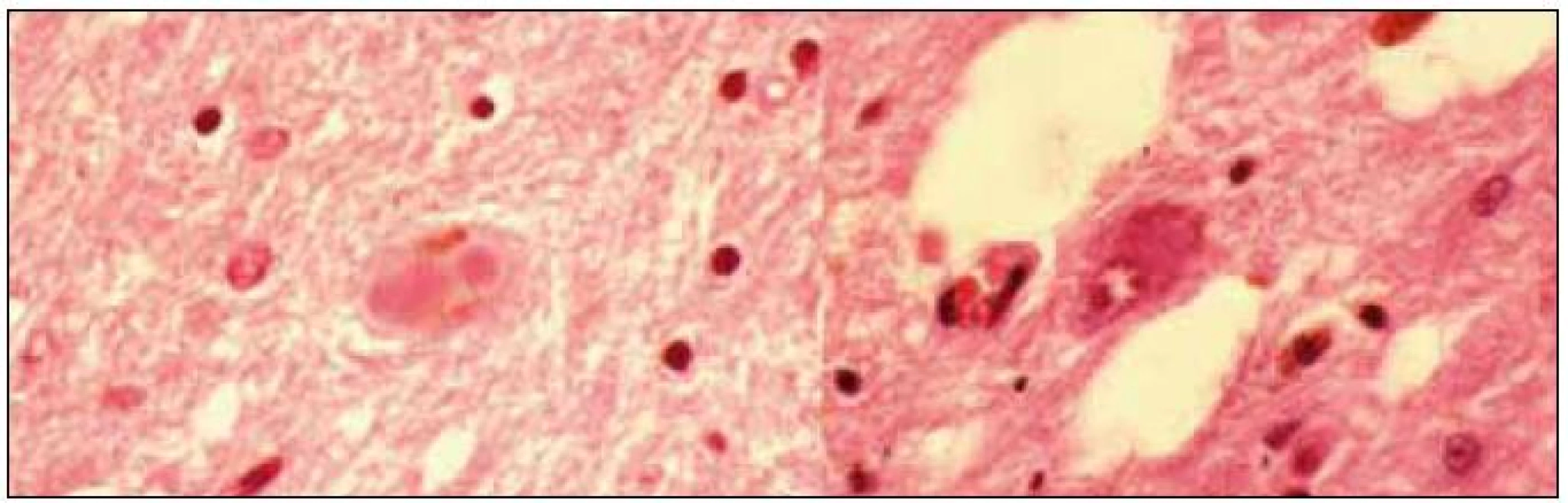
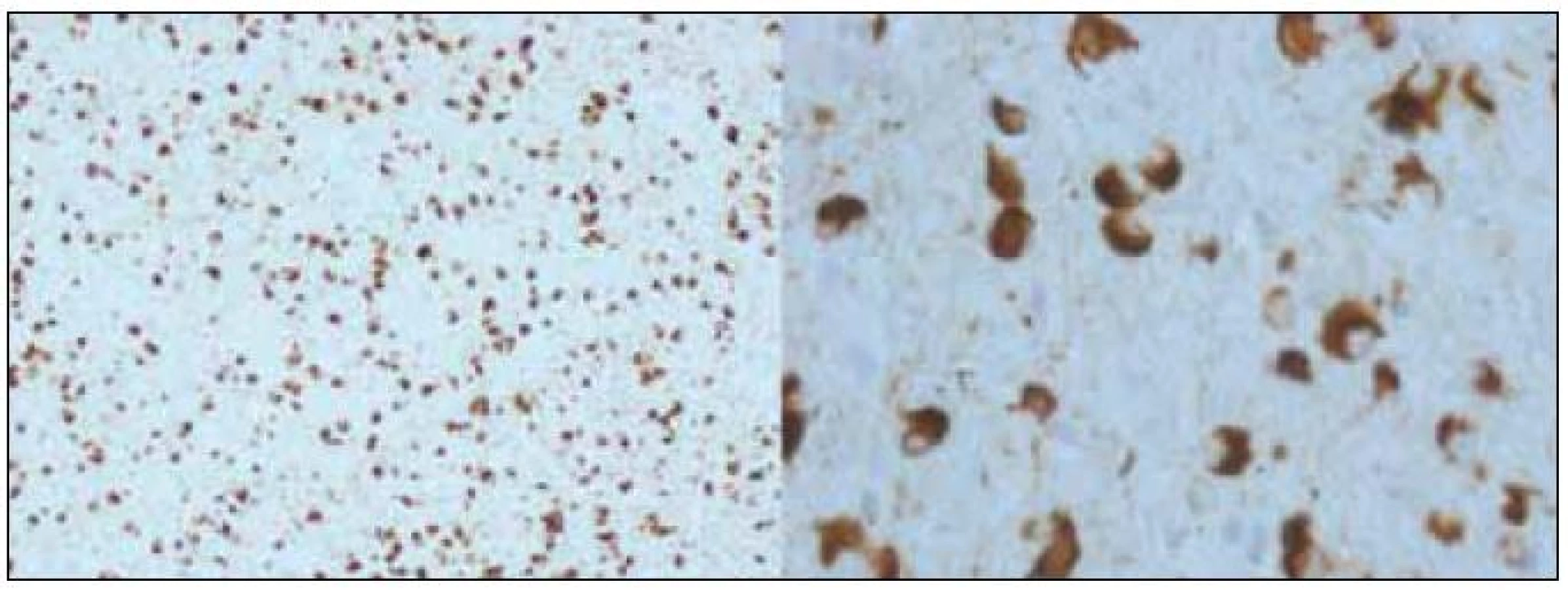
Demence s Lewyho tělísky (DLBD) a Parkinsonova nemoc s demencí (PDD)
Diskuze o incidenci kognitivní poruchy nebo demence u Parkinsonovy nemoci trvá také zhruba 20 let. Původní názor totiž byl, že jen u menšiny pacientů, trpících idiopatickou Parkinsonovou nemocí, se vyvine kognitivní porucha nebo demence. Díky pokrokům v terapii a moderním dopaminergním preparátům, kterými jsou léčeni prakticky všichni pacienti, je přežití pacientů v posledních dekádách významně delší. Vyšlo tím najevo, že u řady z nich se s progresí nemoci vyvíjí i kognitivní deficit, což bylo reflektováno i v publikovaných studiích; při důkladnějším zkoumání větších kohort, které probíhalo zhruba od poloviny 90. let, bylo zjištěno, že až 50 % pacientů trpělo různě vyjádřeným kognitivním deficitem [16]. Toto procento bylo v textech publikovaných v průběhu posledních 10 let průběžně zvyšováno až na více než 90 %, než se nakonec na základě recentních epidemiologických studií ustálilo mezi 30–40 % [17–19]. Ale již v počátcích zkoumání větších kohort bylo patrné, že část těchto pacientů manifestuje distinktní fenotyp, který sdílí mnohé znaky s Alzheimerovou nemocí. Ve stejné době specialisté věnující se Alzheimerově nemoci, např. Laura Fratiglioni a její tým (jako první) popisovali až u 20 % pacientů s Alzheimerovou chorobou extrapyramidové příznaky upomínající nápadně na Parkinsonovu nemoc [20]. Zjištěné skutečnosti se staly podkladem (dodnes nekončící) diskuze, zda skutečně může existovat něco jako „alzheimerská varianta Parkinsonovy nemoci“ nebo „parkinsonská varianta Alzheimerovy nemoci“. Zároveň s touto diskuzí probíhaly klinickopatologické studie, které zjistily, že obě výše uvedené varianty neurodegenerativních onemocnění sdílejí společnou patologii, a to difuzní přítomnost Lewyho tělísek v kortexu i jiných oblastech šedé hmoty mozkové, a zároveň přítomnost neurofilamentových změn a amyloidových plak [21]. V roce 1996 potom Ian McKeith publikoval (dosud jediná) klinická diagnostická kritéria této nemoci, kterou nazval „diffuse Lewy body disease“, a které se dnes říká „Dementia with Lewy bodies (DLBD)“neboli česky „demence s Lewyho tě-lísky“ [22].
![Příloha 3. Klinická diagnostická kritéria demence s Lewyho tělísky [70].](https://pl-master.mdcdn.cz/media/image/cc1c5cb3f2a0f7ec5d60417affe4ce18.jpeg?version=1537790321)
Dnes již víme, že nejčastější fenotyp DLBD je parkinsonský, s absencí třesu. Od klasické Parkinsonovy nemoci (kromě absence třesu, která však může být i u Parkinsonovy nemoci) se odlišuje tím, že pacienti trpí zrakovými halucinacemi a poměrně brzy se u nich vyvine progredující kognitivní porucha, jež (většinou do dvou let) vede k invalidizující demenci. Dalším typickým znakem je vysoká senzitivita vůči neuroleptikům a některým antiemetikům (tietylperazin nebo metoklopramid), jejichž antidopaminergní a anticholinergní efekt může způsobit rozvoj katatonního stavu a vystupňovat svalovou rigiditu. Bohužel se demence s Lewyho tělísky fenotypicky příliš neodlišuje od Parkinsonovy nemoci s demencí (PDD), což je dodnes zdrojem diagnostické i klasifikační konfuze [23]. Existuje nepříliš zřetelná dělicí linie, která odlišuje demenci s Lewyho tělísky od Parkinsonovy nemoci na základě rychlosti, s jakou se vyvine kognitivní porucha na úrovni demence, v současnosti je uznávána arbitrární hranice 12 měsíců. V době ultrastrukturálních a genetických podkladů klasifikace chorobných stavů je to pochopitelně zcela nedostatečné kritérium, ale in vivo nic víc k dispozici není. Jednoznačná diagnostická kritéria Parkinsonovy nemoci s demencí v současnosti neexistují, a je tedy při diferenciální diagnóze nutno vycházet ze stále platných McKeithových kritérií DLBD z roku 1996.
![Příloha č. 4. Klinická diagnostická kritéria Parkinsonovy nemoci s demencí [71].](https://pl-master.mdcdn.cz/media/image/56f04e65e4f2305218c01d922bb913db.jpeg?version=1537790321)
Kortikobazální syndromy (CBS)
Jen krátkou dobu před Quinnovým popisem a definicí multisystémové atrofie se v literatuře začaly objevovat častější popisy pacientů, kteří trpěli parkinsonizmem s nápadným jednostranným postižením [24]. Klasická Parkinsonova nemoc sice většinou (nebo téměř vždy) začíná jako jednostranný syndrom, během prvních let průběhu nemoci ale dochází u naprosté většiny pacientů k symetrizaci, která sice nebývá dokonalá, ale (snad kromě třesu, jenž v řadě případů zůstává akcentovaný na jedné či druhé straně těla) postižení je na obou stranách těla přinejmenším srovnatelné. U popisovaných případů však významná lateralizace symptomů (nikoliv tedy stranová predilekce) zůstávala patrná po celou dobu průběhu jejich nemoci a s progresí se zvýrazňovala. Při bližším zkoumání a opakovaných vyšetřeních bylo záhy jasné, že u takto postižených pacientů je přítomna další porucha, jejíž příčinu je třeba hledat v lézi postihující identickou hemisféru, tj. tu, která odpovídá straně parkinsonského postižení. V rámci metaanalýzy kazuistik (tehdy celkem sedm) bylo zjištěno, že tato porucha byla lokalizována do oblasti mozkové kůry a že je pravděpodobně identická s poruchou, popsanou Rebeizem v roce 1967 po názvem „corticodentatonigral degeneration with neuronal achromasia“. K popisu syndromu byl tedy navržen termín kortikobazální degenerace [25]. Poruchou přídatnou k parkinsonizmu může být u tohoto onemocnění prakticky cokoli: myoklonus, poruchy symbolických funkcí, jako je řeč, porozumění řeči, psaní, čtení, pravolevá a prostorová orientace, může být přítomna porucha nazývaná „alien hand“, tj. syndrom cizí ruky, kdy si končetina „dělá prakticky, co chce“ nezávisle na vůli pacienta [26]. Kortikobazální degenerace je krásný příklad toho, jak konsekutivně dochází ke štěpení určitého parkinsonského fenotypu. V průběhu minulých 20 let bylo postupně zjištěno, že se jedná o další tzv. tauopatii a že je mnohem častější, než se zdálo; zároveň bylo při klinickopatologickém zkoumání zjištěno, že její fenotyp je více než variabilní, neboť dokáže imitovat prakticky jakýkoliv parkinsonský syndrom a je patrně nejčastější příčinou diagnostických omylů [27–29]. Korelační ultrastrukturální studie, publikované zcela recentně, navíc nasvědčují tomu, že kortikobazální degenerace je extrémně heterogenní jednotkou nejen v klinické, ale i v patologické podobě. V řadě případů pacienti jevili klinicky jasný obraz kortikobazální degenerace, nicméně post mortem byly zjištěny patologické změny odpovídající nejenom klasické CBD, ale i Alzheimerově nemoci, FTD nebo PSP. Je tudíž zřejmé, že z klinického obrazu nelze ani minimálně spolehlivě odhadnout charakter odpovídajících patologických změn [30]. Z toho důvodu je v dnešní době termín „kortikobazální degenerace“ rezervován až pro diagnózu patologickou, zatím co všechny klinické manifestace odpovídající původnímu označení CBD jsou souhrnně označovány jako kortikobazální syndromy [31].
![Příloha 5. Klinická diagnostická kritéria kortikobazálního syndromu [72].](https://pl-master.mdcdn.cz/media/image/c6aaa54e03a64967c24de35cc1a83006.jpeg?version=1537790321)
Frontotemporální demence (FTD)
Ještě složitější je situace s tzv. frontotemporální demencí. Toto onemocnění bylo známo více než 100 let jako Pickova demence, a to podle autora prvního popisu onemocnění v roce 1892, dlouholetého přednosty německé neuropsychiatrické kliniky Karlo-Ferdinandovy univerzity v Praze Arnolda Picka, který se narodil ve Velkém Meziříčí jako syn uzenáře; potomci jeho bratra dodnes vyrábějí v Szegedu slavný Pick Szálami, Pickův uherák [32]. Pickova demence byla považována za jeden z typů presenilní demence, aniž se (a to po dobu celých sta let) uvažovalo o nějakém vztahu tohoto onemocnění k extrapyramidovému systému. Pick v podstatě popsal dvě různé manifestace onemocnění, jednu s převažujícími behaviorálními změnami a druhou s převažující poruchou řeči. Všiml si velmi často přítomné klinickoanatomické korelace obou forem s nápadnou atrofií frontálního nebo temporálního laloku [33]. Histologické abnormality související s Pickovou chorobou nepopsal Pick sám, ale až v roce 1911 Alois Alzheimer (obr. 4). Zhruba od 20. let minulého století se vžil a začal užívat eponym Pickova choroba, která byla považována za jednu klinickou entitu, v jejímž průběhu se rozlišovaly tři fáze: první s poruchami chování, druhá s fokálními symptomy v podobě afázie a třetí s generalizovanou demencí [34]. Nejprve Sanders v roce 1939 a následně Schenk v roce 1951 popsali manifestaci tohoto onemocnění ve velké německé rodině [35]. Dědičnost frontotemporální demence s vazbou na chromozom 17 však byla dokumentována až v roce 1994 [36]. Mezitím ovšem proběhla řada klinickopatologických studií, na základě jejichž výsledků prošla terminologie tohoto onemocnění několika fázemi. Od „demence frontálního typu“ [37] přes „frontální degeneraci“ [38] nebo „frontální degeneraci non-alzheimerovského typu“ [39] či „frontotemporální demenci“ až po současné označení. Dnes uznávané označení, vycházející jak z klinické manifestace, tak z klinicko-anatomicko-patologických korelací degenerativních změn, je FTLD (Frontotemporal Lobar Degeneration) s třemi subtypy: bvFTD – behaviorální varianta bez zjevného jazykového deficitu, sémantická demence (SD) a progresivní non-fluentní afázie, PNFA [40]. Na základě nejnovějších poznatků se však zdá, že ani toto označení nebude konečné [41].
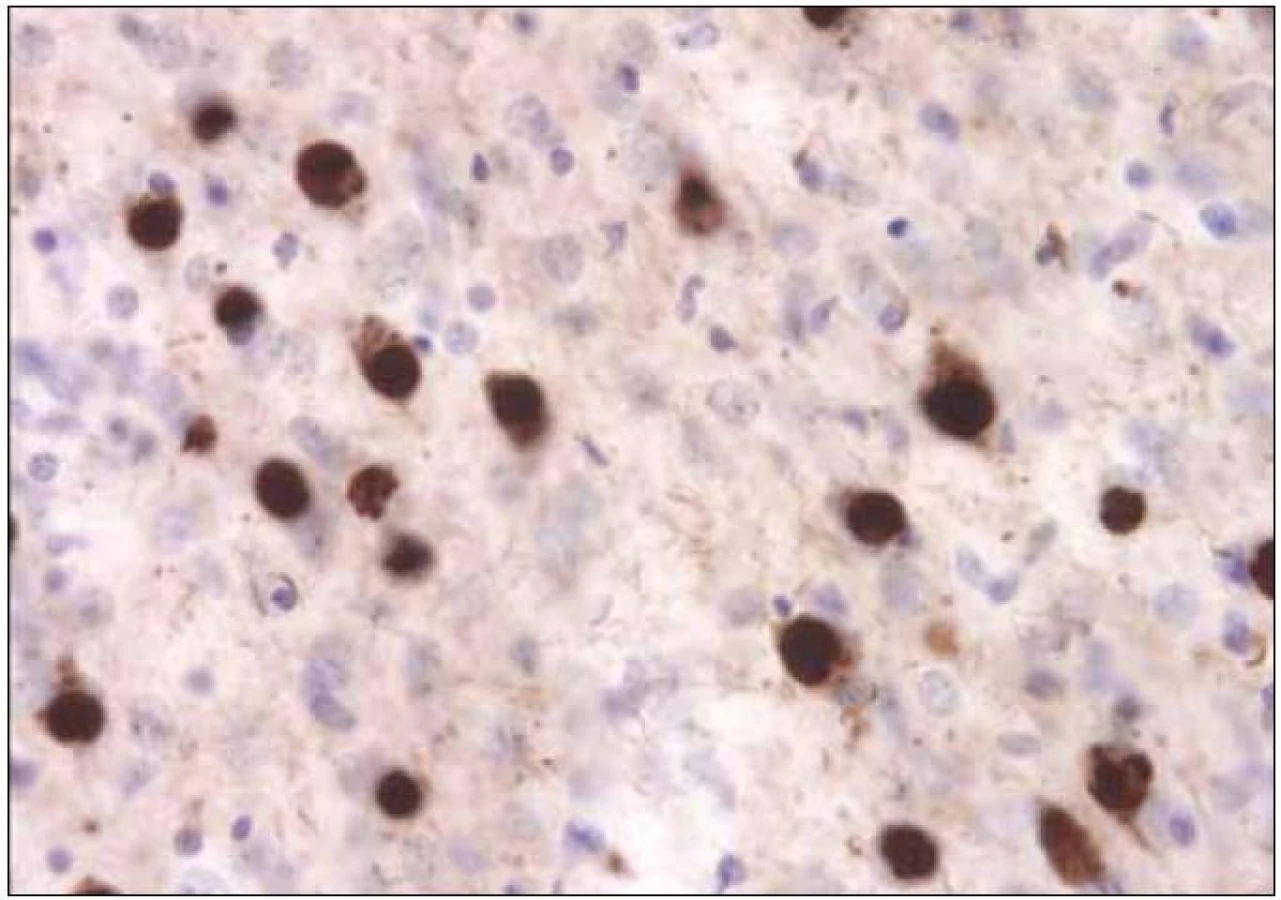
![Příloha 6. Klinická diagnostická kriteria behaviorální varianty frontotemporální demence (bvFTD) [73].](https://pl-master.mdcdn.cz/media/image/433900871de2204cf7462a5c62237534.jpeg?version=1537790321)
![Příloha 7. Klinická diagnostická kritéria progresivní non-fluentní afázie jako formy frontotemporální demence (FTLD-PNFA) [73].](https://pl-master.mdcdn.cz/media/image/6f877d7d88bac8c1407a62413f1460ad.jpeg?version=1537790321)
![Příloha 8. Klinická diagnostická kritéria sémantické demence jako formy frontotemporální demence (FTLD-SD) [73].](https://pl-master.mdcdn.cz/media/image/e0762425335befb0205c1a1d50d80ed5.jpeg?version=1537790321)
![Příloha 9. Kritéria společná pro všechny varianty frontotemporální demence (FTLD) [73].](https://pl-master.mdcdn.cz/media/image/cd531bddb50b7953872ee7c557520432.jpeg?version=1537790321)
„Neurodegenerative overlap syndrome“ a guamský parkinsonský komplex
Asi před 15 lety se začala častěji objevovat sdělení uvádějící, že někteří pacienti trpící frontotemporální demencí jeví i znaky parkinsonského fenotypu. U řady pacientů v takovém případě rozvoj symptomů FTLD předchází rozvoj kortikobazálního syndromu a vice versa a konečnou diagnózou patologickou je většinou CBD. O něco méně častěji se manifestuje současně s FTLD i PSP fenotyp [42]. Nicméně u řady pacientů manifestujících FTLD nejsou přítomny jen příznaky parkinsonské, ale i příznaky amyotrofické laterální sklerózy, dalšího neurodegenerativního onemocnění, které však ve své původní podobě postihuje především míchu. To už začalo být hodně zajímavé, a to pro nápadnou podobnost tohoto syndromu s tím, čemu se říká „lytico-bodig“ v chamorrštině, a tzv. guamský parkinsonský komplex v odborné literatuře. Jde o endemickou nemoc vyskytující se pouze na ostrově Guam v Marianách a na poloostrově Kii ostrova Kjúšú v Japonsku. Nemoc byla poprvé popsána španělskými misionáři před více než 200 lety (jde tedy o popis starší než Parkinsonův), a byla na uvedených místech poměrně (až velmi) často přítomna do 70. let minulého století, kdy její incidence začala pozvolna klesat. Nemoc byla charakteristická kombinací parkinsonizmu s amyotrofickou laterální sklerózou (ALS) a progredující demencí. Příčina nemoci byla dlouho neznámá. Mluvilo se o pomalých virech apod., ale nakonec bylo s určitou pravděpodobností zjištěno, že příčinou nemoci může být chronická intoxikace, jež vzniká požíváním masa malých netopýrů (tzv. Mariana fruit bat), kteří se téměř výlučně živí šťávou z cykasových plodů (obr. 5). A právě tato šťáva obsahuje specifický neurotoxin, který způsobuje pozvolné odumírání a zánik určitých nervových buněk [43]. Tím se dostáváme obloukem k osobě Johna Steelea, protože právě on to byl, kdo se se svým týmem dopátral celého potravního řetězce, jenž může vést k rozvoji guamského parkinsonského komplexu (přestože i tato hypotéza je některými kolegy zpochybňována).

Jak velké tedy muselo být překvapení specialistů, kteří v posledních zhruba 15 letech opakovaně ve svých ordinacích ve Spojených státech i Evropě viděli pacienty, kteří se až neuvěřitelně nápadně podobali těm z ostrova Guam? Pacienti několik let léčeni pro klasickou Parkinsonovou nemoc, a náhle se u nich objevily příznaky amyotrofické laterální sklerózy a progredující demence? V první chvíli nebylo jasné, zda nejde o diagnostické omyly, ale skutečně se jednalo o pacienty, kteří nadále dobře odpovídali na léčbu L-DOPA, jejich demence progredovala a postupně přestali být schopni mluvit a polykat [44–46]. Tedy jasný obraz guamského parkinsonského komplexu, o kterém však Steele poměrně přesvědčivě tvrdí, že je exotoxického původu. Zatím medicínská záhada, jejíž vysvětlení dosud neexistuje a nemá smysl se pouštět do spekulací, neboť dat je v tuto chvíli relativně málo a musíme pečlivě zkoumat všechny okolnosti, než vyslovíme alespoň hypotézu. Je však jasné, že existuje další parkinsonský fenotyp, který se pro nedostatek trefnější terminologie v současnosti nazývá již před 15 lety použitým termínem „neurodegenerative overlap syndrome“, tedy syndrom překrývající se neurodegenerace [47].
Guadeloupský parkinsonizmus
Znalost guamského parkinsonského komplexu pravděpodobně způsobila, že bližší poznání a popis dalšího endemického atypického parkinsonského syndromu trval mnohem kratší dobu. Jedná se o tzv. guadeloupský parkinsonizmus, který kombinuje klasické parkinsonské příznaky a demenci, ve více než v polovině případů doplněné symptomy typickými spíše pro PSP, jako je posturální instabilita a pády, okohybné poruchy, dysartrie a patologické kožní reflexy [48]. Podobně jako u guamského komplexu i v případě guadeloupského parkinsonizmu byla hledána ekotoxická noxa. Názor se nakonec ustálil na tzv. charlotinkách („sourcrop“ ve francouzské kreolštině), plodech rostliny z čeledi Anonnaceae (Annona muricata), které jsou na ostrově hustě konzumovány (obr. 6). Bylo zjištěno, že obsahují specifický neurotoxin z třídy acetogeninů. Předpokládá se však nepochybná familiární susceptibilita, a to vzhledem k tomu, že prevalence onemocnění není nijak dramaticky vysoká, přičemž konzumaci zmíněných plodů se oddává téměř celá ostrovní populace [49].

Brait-Fahnova nemoc
Další extrémně vzácná koexistence parkinsonizmu a onemocnění motoneuronu v podobě ALS byla poprvé popsána před téměř 30 lety Kennethem Braitem a Stanley Fahnem [50,51] Na rozdíl od guamského parkinsonského komplexu s příznačnou demencí a na rozdíl od multisystémové atrofie s autonomními nebo cerebelárními projevy nedominují v tomto případě žádné další neurologické příznaky. Doposud byl ALS-parkinsonizmus (ALS-P nebo též „Brait-Fahn disease“) popsán u 42 pacientů, z nichž u téměř poloviny byla zaznamenána dobrá odpovídavost na L-DOPA [52–54]. Přesný patofyziologický mechanizmus nebyl jednoznačně objasněn, ale některé studie poukazují na překrývání obou jednotek nejen v rovině klinického obrazu, ale i ve výsledcích epidemiologických šetření, neurozobrazovacích vyšetření či patologických nálezů. Radioizotopové zobrazovací metody poukázaly na dopaminergní deficit u pacientů s ALS i bez známek parkinsonizmu [55]. Častější výskyt Parkinsonovy nemoci byl prokázán i u příbuzných pacientů s ALS [52]. Není překvapením, že u pacienta s typickou dopa-responzivní Parkinsonovou nemocí bez demence a bez známek onemocnění motoneuronu byla popsána těžká degenerace nigrálních neuronů, ale nikoli s přítomností Lewyho tělísek, nýbrž s patologickým nálezem shodným s ALS [56]. Obdobně je popsána degenerace nigrálních neuronů s Lewyho tělísky u pacientů se sporadickou ALS, ale bez jakýchkoliv parkinsonských příznaků [57]. Patologicky byla dokumentována u ALS-P současná přítomnost jak typického nálezu pro Parkinsonovu nemoc s Lewyho tělísky a degenerací dopaminergních neuronů, tak typického nálezu pro ALS s numerickou atrofií velkých motorických neuronů předních rohů míšních a nálezem eosinofilních granulárních inkluzí tzv. Buninových tělísek a inkluzí reagujících na ubikvitinová antiséra. Nálezy těchto studií naznačují, že amyotrofická laterální skleróza i Parkinsonova nemoc by mohly sdílet obdobné patogenetické mechanizmy, ale žádná společná mutace zatím popsána nebyla. Kromě ALS-P, multisystémové atrofie, guamského komplexu a FTLD-parkinsonizmus-amyotrofie vázané na chromozom 17 se onemocnění motoneuronu kombinované s parkinsonizmem nachází i u další neurodegenerativních onemocněních. Příkladem je primární laterální skleróza-parkinsonizmus nebo komplikovaná hereditární spastická paraparéza. U typu SPG10 a SPG11 je parkinsonizmus první manifestací onemocnění a až v pozdějším průběhu se přidávají příznaky postižení horního motoneuronu v podobě spastické paraplegie [50,54].
Spinocerebellární ataxie (SCA)
Nakonec je třeba se zmínit o parkinsonském fenotypu, který se manifestuje společně s dominantním mozečkovým syndromem. Tento obraz je charakteristický pro spinocerebellární ataxie, skupinu onemocnění, u nichž dochází k postupně progredující mozečkové degeneraci, postižení aferentních a eferentních spojení mozečku, zadních provazců míšních, případně pyramidové dráhy, pontinních jader a jiných částí CNS. V minulosti byly mozečkové degenerace velmi málo prozkoumanou oblastí, u níž výrazný pokrok nastal až v 90. letech minulého století v souvislosti s expanzí molekulární biologie a genetiky. Byla objevena řada genů zodpovědných za klinické příznaky, objevila se možnost stanovení přesné diagnózy a in-vivo lokalizace lokusu, chromozomu, genu a genového produktu. Spinocerebellární ataxie jsou charakterizovány širokým spektrem neurologických příznaků: kromě dominujících symptomů odpovídajících palleocerebellárnímu a neocerebellárnímu syndromu se mohou objevit extracerebellární příznaky, jako např. atrofie optiku, katarakta, spasticita, z poruch extrapyramidových především parkinsonizmus, ale také dystonie, myoklonus, třes, nebo chorea; s parkinsonizmem koexistují periferní neuropatie (typická je axonální neuropatie), sfinkterové poruchy, postižení kognitivních funkcí až charakteru demence, rozhodovacích procesů či epilepsie [58]. Dominantní je však u všech typů SCA postižení mozečku, struktury po dlouhá desetiletí opomíjené zejména ve vztahu ke kognitivním a non-motorickým funkcím [58–61]. Nejvíce prozkoumanou skupinou SCA jsou autozomálně dominantní SCA (AD-SCA), u nichž je v současné době registrováno 28 prokázaných genetických typů. Incidence se odhaduje na 3/100 000 a je zjevný rozdíl v geografické distribuci [62]. Klinická diagnóza specifických subtypů AD-SCA je ovšem významně komplikována výrazným překrýváním fenotypických příznaků mezi jednotlivými genotypy a výraznou variabilitou klinických příznaků v jednotlivých genotypech AD-SCA [63]. Velmi výrazný parkinsonský fenotyp se může objevit u SCA 2, SCA 3 (nejčastější AD-SCA v Evropě, velmi častá např. v Brazílii) či velmi vzácně se vyskytujících SCA 12 a SCA 21. Byly popsány abnormity při DaTSCAN („dopamine transporter imaging“) vyšetření s predominantním postižením presynaptické dopaminergní funkce u různých typů AD-SCA, např. SCA 2,3 [64]. Patologické studie prokázaly poměrně široké spektrum neurodegenerace: ztráty neuronů, atrofie a gliové změny v oblasti substantia nigra, nucleus caudatus, globus pallidus, putamen, nucleus subthalamicus, tedy prakticky všech jader extrapyramidového systému u řady typů AD-SCA (např. SCA 1,2,3,6,7,17, a dentato-rubro-pallido-luysiánské atrofie, tzv. DRPLA [62]. Recentně publikovaná práce zdůrazňuje přítomnost extrapyramidových příznaků a SCA 2 genotyp jako důležité negativní aspekty signifikantně zvyšující pády u pacientů s SCA [65]. U SCA s dominantním parkinsonským fenotypem dokonce dobře funguje dopaminergní léčba a v literatuře se objevují zmínky o symptomatickém účinku amantadinu u SCA3 nebo hluboké mozkové stimulace u SCA2 [66]. Spektrum diferenciální diagnostiky atypických parkinsonských syndromů rozšiřuje (a pochopitelně komplikuje) syndrom fragilního chromozomu X s tremorem/ataxií (FXTAS), který je vázán na dospělý věk a může se projevit kromě tremoru, ataktické chůze, autonomní dysfunkce a neuropatií také atypickým parkinsonským syndromem a demencí [67].
Závěr
Medicína je v zásadě biologická věda a jako ostatní biologické vědy i ona se rozvíjí na základě dalšího poznání ultrastrukturální morfologie a jejích souvislostí. I vývoj poznání parkinsonské neurodegenerace v posledních 100 letech je v podstatě reflexí zlepšeného klinického pozorování déle přežívajících pacientů a rozmachu morfologických a genetických metod. Dnešní Parkinsonova nemoc je prostě jinou nemocí než nemoc, kterou popsal James Parkinson, a dnešní parkinsonský fenotyp, ať už je jakéhokoliv druhu, je ze všeho nejvíce věrným odrazem progredující neurodegenerace. Ta může mít přímou, kaudokraniální progresi, kdy je degenerativním procesem zachvacována postupně další a další část mozku ve směru od mozkového kmene k hemisférám. Potom se onemocnění bude lékařům a okolí jevit jako nemoc velmi blízká Parkinsonovu popisu. Jakákoliv „odbočka“ z této relativně přímé cesty neurodegenerace však bude příčinou toho, že se rozvine jiný fenotyp než onen klasický. Vyvine se některý z výše popsaných fenotypů, které pro nedostatek lepší terminologie nazýváme „atypické“ a které v současnosti klinicky pouze nedokonale charakterizujeme za pomoci (bez výjimky) obsolentních a většinou prakticky nepoužitelných klinických diagnostických kritérií (přílohy 1–9) [68–73].
Bližší (a doufejme že skutečné) vztahy mezi klasickým obrazem onemocnění a obrazy atypickými a mezi charakterem a místem neurodegenerace zatím mapujeme [74]; do jaké míry se na klinickém fenotypu projevuje dominantní typ neurodegenerace, tj. tauopatie nebo alfa-synukleinopatie a jejich vzájemný poměr či překrývání, je v současné době jen luštěnou záhadou [75].
Poděkování
Práce byla podpořena grantovým projektem IGA UP LF_2011_012 a grantem IGA MZ ČR NT12221-5 (K. Farníková, P. Kaňovský).
MUDr. Kateřina Farníková
Neurologická klinika LF UP a FN Olomouc
I. P. Pavlova 6
775 20 Olomouc
e-mail: katmen@centrum.cz
Přijato k recenzi: 28. 2. 2011
Přijato do tisku: 27. 6. 2011
Sources
1. Parkinson J. An essay on the shaking palsy. London: Sherwood, Neely & Jones 1817.
2. Reichmann H. Clinical criteria for the diagnosis of Parkinson‘s disease. Neurodegener Dis 2010; 7(5): 284–290.
3. Marras C, Lang A. Invited article: changing concepts in Parkinson disease: moving beyond the decade of the brain. Neurology 2008; 70(21): 1996–2003.
4. Song YJ, Huang Y, Halliday GM. Clinical correlates of similar pathologies in parkinsonian syndromes. Mov Disord 2011; 26(3): 499–506.
5. Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A et al. Mutation in the alpha-synuclein gene identified in families with Parkinson‘s disease. Sicence 1997; 276: 2045–2047.
6. Shulman JM, DeJager PL, Feany MB. Parkinson‘s disease: genetics and pathogenesis. Annu Rev Pathol 2010; 6: 193–222.
7. Richardson JC, Steele J, Oslzewski J. Supranuclear ophtamoplegia, pseudobulbar palsy, nuchal dystonia and dementia. A clinical report on eight case sof „heterogenous system degeneration“. Trans Am Neurol Assoc 1963; 88: 25–29.
8. Steele JC, Richardson JC, Olszewski J. Progressive supranuclear palsy: a heterogeneous degeneration involving the brain stem, basal ganglia and cerebellum with vertical gaze and pseudobulbar palsy, nuchal dystonia and dementia. Arch Neurol 1964; 10: 333–359.
9. Williams DR, Holton JL, Strand K, Revesz T, Lees AJ. Pure akinesia with gait freezing: a third clinical phenotype of progressive supranuclear palsy. Mov Disord 2007; 22: 2235–2241.
10. Steele JC. PSP Steele-Richardson-Olszewski syndrome Richardson‘s disease. J Neurol Sci 2009; 285 (Suppl 1): S25.
11. Schwarz GA. The orthostatic hypotension syndrome of Shy-Drager. A clinicopathologic report. Arch Neurol 1967; 16: 123–139
12. Quinn N. Multiple system atrophy – the nature of the beast. J Neurol Neurosurg Psychiatry 1989; 52 (Suppl): 78–89.
13. Kaňovský P, Streitová H, Bareš M, Kuba R, Pospíšilová D. Multisystémová atrofie – nový nosologický koncept. Cesk Slov Neurol Neurochir 1996; 59: 3–9.
14. Stefanova N, Bücke P, Duerr S, Wenning GK. Multiple system atrophy: an update. Lancet Neurol 2009; 8(12): 1172–1178.
15. Fearnley JM, Lees AJ. Striatonigral degeneration. A clinicopathological study. Brain 1990; 113 (Pt 6): 1823–1842.
16. Aarsland D, Zaccai J, Brayne C. A systematic review of prevalence studies of dementia in Parkinson‘s disease. Mov Disord 2005; 20(10): 1255–1263.
17. Riedel O, Klotsche J, Spottke A, Deuschl G, Förstl H, Henn F et al. Cognitive impairment in 873 patients with idiopathic Parkinson‘s disease. Results from the German Study on Epidemiology of Parkinson‘s disease with Dementia (GEPAD). J Neurol 2008; 255(2): 255–264.
18. Aarsland D, Kurz MW. The epidemiology of dementia associated with Parkinson disease. J Neurol Sci 2010; 289(1–2): 18–22
19. Von Reichmann H, Deuschl G, Riedel O, Spottke A, Förstl H, Henn F et al. The German Study on the Epidemiology of Parkinson‘s Disease with Dementia (GEPAD): more than Parkinson. MMW Fortschr Med 2010; 152 (Suppl 1): 1–6.
20. Torres HA, Fratiglioni L, Hofman W, Winblad B. Early symptoms and neurological findings in demented subjects from a community survey. Alzheimer Dis Assoc Disord 1995; 9(3): 170–175.
21. Victoroff J, Mack WJ, Lyness SA, Chui HC. Multicenter clinicopathological correlation in dementia. Am J Psychiatry 1995; 152(10): 1476–1484.
22. McKeith IG, Galasko D, Kosaka K, Perry EK, Dickson DW, Hansen LA et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology 1996; 47(5): 1113–1124.
23. Mollenhauer B, Förstl H, Deuschl G, Storch A, Oertel W, Trenkwalder C. Lewy body and parkinsonian dementia: common, but often misdiagnosed conditions. Dtsch Arztebl Int 2010; 107(39): 684–691.
24. Gibb WR, Luthert PJ, Marsden CD. Corticobasal degeneration. Brain 1989; 112 (Pt 5): 1171–1192.
25. Rebeisz JJ, Kolodny EH, Richardson EP Jr. Corticodentatonigral degeneration with neuronal achromasia: a progressive disorder of late adult life. Trans Am Neurol Assoc 1967; 92: 23–26.
26. Lang AE. Parkinsonism in corticobasal degeneration. Adv Neurol 2000; 82: 83–89.
27. Litvan I, Grimes DA, Lang AE. Phenotypes and prognosis: clinicopathological studies of corticobasal degeneration. Adv Neurol 2000; 82: 183–196.
28. Doran M, du Plessis DG, Enevoldson TP, Fletcher NA, Ghadiali E, Larner AJ. Pathological heterogenity of clinically diagnosed corticobasal degeneration. J Neurol Sci 2003; 216(1): 127–134.
29. Wadia PM, Lang AE. The many faces of corticobasal degeneration. Parkinsonism Relat Disord 2007; 13 (Suppl 3): S336–S340.
30. Ling H, O‘Sullivan SS, Holton JL, Revesz T, Massey LA, Williams DR et al. Does corticobasal degeneration exist? A clinicopathological re-evaluation. Brain 2010; 133 (Pt 7): 2045–2057.
31. Strupp M. Corticobasal syndrome: a field of uncertainty. J Neurol 2011; 258(1): 173–175.
32. Berrios GE, Girling DM. Introduction: Pick‘s disease and the ‚frontal lobe‘ dementias. Hist Psychiatry 1994; 5 (20 Pt 4): 539–547.
33. Pick A, Girling DM, Berrios GE. On the symptomatology of left-sided temporal lobe atrophy. Classic Text No. 29. (Translated and annotated by D.M. Girling and G.E. Berrios.) Hist Psychiatry 1997; 8 (296 pt 1): 149–159.
34. Girling DM, Berrios GE. On the relationship between senile cerebral atrophy and aphasia. Hist Psychiatry 1994; 5: 542–547.
35. Schenk VW. Re-examination of a family with Pick‘s disease. Ann Hum Genet 1959; 23: 325–333.
36. Lynch T, Sano M, Marder KS, Bell KL, Foster NL, Defendini RF et al. Clinical characteristics of a family with chromosome 17-linked disinhibition-dementia-parkinsonism-amyotrophy complex. Neurology 1994; 44(10): 1878–1884.
37. Neary D, Snowden JS, Bowen DM, Sims NR, Mann DM, Yates PO et al. Cerebral biopsy in the investigation of presenile dementia due to cerebral atrophy. J Neurol Neurosurg Psychiatry 1986; 49(2): 157–162.
38. Miller BL, Cummings JL, Villanueva-Meyer J, Boone K, Mehringer CM, Lesser IM et al. Frontal lobe degeneration: Clinical, neuropsychological and SPECT characteristics. Neurology 1991; 41(9):1374–1382.
39. The Lund and Manchester Groups. Clinical and neuropathological criteria for frontotemporal dementia. J Neurol Neurosurg Psychiatry 1994; 57: 416–418.
40. Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 1998; 51(6): 1546–1554.
41. Rohrer JD, Geser F, Zhou J, Gennatas ED, Sidhu M, Trojanowski JQ et al. TDP-43 subtypes are associated with distinct atrophy patterns in frontotemporal dementia. Neurology 2010; 75(24): 2204–2211.
42. Foster NL, Wilhelmsen K, Sima AA, Jones MZ, D‘Amato CJ, Gilman S. Frontotemporal dementia and parkinsonism linked to chromosome 17: a consensus conference. Ann Neurol 1997; 41(6): 706–715.
43. Steele JC, McGeer PL. The ALS/PDC syndrome of Guam and the cycad hypothesis. Neurology 2008; 70(21): 1984–1990.
44. Wszolek ZK, Vieregge P, Uitti RJ, Gasser T, Yasuhara O, McGeer P et al. German-Canadian family (family A) with parkinsonism, amyotrophy, and dementia – Longitudinal observations. Parkinsonism Relat Disord 1997; 3(3): 125–139.
45. Imamura A, Wszolek Z, Uitti R. Neurodegenerative overlap syndrome: parkinsonism and motor neuron disorder. Mov Disord 2007; 22(1): 151–152.
46. Farníková K, Kanovský P, Nestrasil I, Otruba P. Coexistence of parkinsonism, dementia and upper motor neuron syndrome in four Czech patients. J Neurol Sci 2010; 296(1–2): 47–54.
47. Uitti RJ, Berry K, Yasuhara O, Eisen A, Feldman H, McGeer PL et al. Neurodegenerative ‚overlap‘ syndrome: Clinical and pathological features of Parkinson‘s disease, motor neuron disease, and Alzheimer‘s disease. Parkinsonism Relat Disord 1995; 1(1): 21–34.
48. Caparros-Lefebvre D, Lees AJ. Atypical unclassifiable parkinsonism on Guadeloupe: an environmental toxic hypothesis. Mov Disord 2005; 20 (Suppl 12): S114–S118.
49. Lannuzel A, Ruberg M, Michel PP. Atypical parkinsonism in the Caribbean island of Guadeloupe: etiological role of the mitochondrial complex I inhibitor annonacin. Mov Disord 2008; 23(15): 2122–2128.
50. Anheim M, Lagier-Tourenne C, Stevanin G, Fleury M, Durr A, Namer IJ et al. SPG11 spastic paraplegia. A new cause of juvenile parkinsonism. J Neurol 2009; 256(1): 104–108.
51. Brait K, Fahn S, , Schwarz GA. Sporadic and familial parkinsonism and motor neuron disease. Neurology 1973; 23(9):990–1002.
52. Fallis BA, Hardiman O. Aggregation of neurodegenerative disease in ALS kindreds. Amyotroph Lateral Scler 2009; 10(2): 95–98.
53. Gilbert RM, Fahn S, Mitsumoto H, Rowland LP. Parkinsonism and motor neuron diseases: twenty-seven patients with diverse overlap syndromes. Mov Disord 2010; 25(12): 1868–1875.
54. Goizet CA, Boukhris A, Mundwiller E, Tallaksen C, Forlani S, Toutain A et al. Complicated forms of autosomal dominant hereditary spastic paraplegia are frequent in SPG10. Hum Mutat 2009; 30(2): E376–E385.
55. Hideyama T, Momose T, Shimizu J, Tsuji S, Kwak S. A positron emission tomography study on the role of nigral lesions in parkinsonism in patients with amyotrophic lateral sclerosis. Arch Neurol 2006; 63(12): 1719–1722.
56. Shintaku M, Oyanagi K, Kaneda D. Amyotrophic lateral sclerosis with dementia showing clinical parkinsonism and severe degeneration of the substantia nigra: report of an autopsy case. Neuropathology 2007; 27(3): 295–299.
57. Kato S, Oda M, Tanabe H. Diminution of dopaminergic neurons in the substantia nigra of sporadic amyotrophic lateral sclerosis. Neuropathol Appl Neurobiol 1993; 19(4): 300–304.
58. Bares M, Lungu O, Liu T, Waechter T, Gomez CM, Ashe J. Impaired predictive motor timing in patients with cerebellar disorders. Exp Brain Res 2007; 180(2): 355–365.
59. Bares M, Lungu OV, Husárová I, Gescheidt T. Predictive motor timing performance dissociates between early diseases of the cerebellum and Parkinson‘s disease. Cerebellum 2010; 9(1): 124–135.
60. Bareš M. Onemocnění mozečku. Neurol pro Prax 2007; 8(5): 267.
61. Bares M, Lungu OV, Liu T, Waechter T, Gomez CM, Ashe J. The neural substrate of predictive motor timing spinocerebellar ataxia. Cerebellum 2011; 10(2): 233–244.
62. Schöls L, Bauer P, Schmidt T, Schulte T, Riess O. Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. Lancet Neurol 2004; 3(5): 291–304.
63. Maschke M, Oehlert G, Xie TD, Perlman S, Subramony SH, Kumar N et al. Clinical feature profile of spinocerebellar ataxia type 1-8 predicts genetically defined subtypes. Mov Disord 2005; 20(11): 1405–1412.
64. Manto MU. The wide spectrum of spinocerebellar ataxias (SCAs). Cerebellum 2005; 4(1): 2–6.
65. Fonteyn EM, Schmitz-Hübsch T, Verstappen CC, Baliko L, Bloem BR, Boesch S et al. Falls in spinocerebellar ataxias: Results of the EuroSCA Fall Study. Cerebellum 2010; 9(2): 232–239.
66. Freund HJ, Barnikol UB, Nolte D, Treuer H, Auburger G, Tass PA et al. Subthalamic-thalamic DBS in a case with spinocerebellar ataxia type 2 and severe tremor-A unusual clinical benefit. Mov Disord 2007; 22(5): 732–735.
67. Zumrová A, Mušová Z, Košťálová E, Apltová L, Křepelová A, Paděrová A. Autozomálně recesivní a X-vázané ataxie. Neurol pro Prax 2007; 8(5): 272–276.
68. Litvan I, Agid Y, Calne D, Campbell G, Dubois B, Duvoisin RC et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steel-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology 1996; 47(1): 1–9.
69. Gilman S, Low PA, Quinn N, Albanese A, Ben-Shlomo Y, Fowler CJ et al. Consensus statement on the diagnosis of multiple system atrophy. J Auton Nerv Syst 1998; 74(2–3): 189–192.
70. McKeith IG, Dickson DW, Lowe J, Emre M, O‘Brien JT, Feldman H et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB consortium. Neurology 2005; 65(12): 1863–1872.
71. Emre M, Aarsland D, Brown R, Burn DJ, Duyckaerts C, Mizuno Y et al. Clinical diagnostic criteria for dementia associated with Parkinson‘s disease. Mov Disord 2007; 22(12): 1689–1707.
72. Boeve BF, Lang AE, Litvan I. Corticobasal degeneration and its relationship to progressive supranuclear palsy and frontotemporal dementia. Ann Neurol 2003; 54 (Suppl 5): S15–S19.
73. Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S et al. Frontotemporal lobar degeneration: A consensus on clinical diagnostic criteria. Neurology 1998; 51(6): 1546–1554.
74. Přikrylová Vránová H, Mareš J, Nevrlý M, Stejskal D, Zapletalová J, Hluštík P et al. CSF markers of neurodegeneration in Parkinson‘s disease. J Neural Transm 2010; 117(10): 1177–1181.
75. Galpern WR, Lang AE. Interface between tauopathies and synucleinopathies: a tale of two proteins. Ann Neurol 2006; 59(3): 449–458.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2011 Issue 6
Most read in this issue
- Miller Fisherův syndrom – čtyři vlastní pozorování a přehled současných poznatků
- Současný pohled na patofyziologii migrény
- Operační léčba poranění plexus brachialis
- Novelizace české verze Addenbrookského kognitivního testu (ACE-CZ)