
Adultní forma glutarové aciduri e II. typu – nedostatečně di agnostikovaná příčina proximální myopati e – kazuistika
Adult Form of Glutaric Aciduri a Type II – Under di agnosed Ca useof Proximal Myopathy – a Case Report
Glutaric aciduri a type II or multiple acyl‑ CoA dehydrogenase defici ency (MIM 231680) is an a utosomal recessively inherited disorder with heterogene o us clinical manifestati on and genetic backgro und, bi ochemically characterized by the accumulati on of specific metabolites so urcing from the defici ent capacity of flavin oxidative enzymes transferring electrons into the system of electron- transfer flavoprotein (ETF) and electron- transfer flavoprotein:ubiquinone oxidoreductase (ETF:QO). Clinical phenotypes include not only the lethal ne onatal forms, but also the late‑onset disorder, which usu ally manifests with proximal myopathic syndrome and sometimes with attacks of hypoketotic hypoglycemi a. The incidence of the myopathic form of glutaric aciduria type II has not been determined; nevertheless, owing to e asi er access to novel di agnostic methods, including tandem mass spectrometry, and the ever incre asing awareness of clinici ans, the number of di agnosed pati ents has been growing constantly. The case report of o ur pati ent, for the first time di agnosed on the DNA level in the Czech Republic, illustrates not only the clinical co urse of the dise ase, but also the possibiliti es of di agnostics and therapy. It might serve as an inspirati on for ne urological practice.
Key words:
glutaric aciduria type II – multiple acyl coenzyme A dehydrogenase deficiency – proximal myopathy – riboflavin
:
H. Jahnová 1,2; P. Chrastina 1; M. Havlová 3; J. Zámečník 4
:
Ústav dědičných metabolických poruch VFN a 1. LF UK v Praze, 2Klinika dětí a dorostu FNKV, Praha, 3Neurologická klinika VFN a 1. LF UK v Praze, 4Ústav patologi e a molekulární medicíny FN Motol a 2. LF UK v Praze
1
:
Cesk Slov Neurol N 2009; 72/105(3): 260-264
:
Case Report
Glutarová aciduri e II. typu neboli mnohočetný deficit acyl‑ CoA dehydrogenáz (MIM 231680) je klinicky i geneticky heterogenní, a utozomálně recesivně dědičná metabolická porucha, bi ochemicky charakterizovaná akumulací specifických metabolitů v důsledku omezené kapacity flavinových dehydrogenáz předávajících elektrony do systému elektrony přenášejícího flavoproteinu (ETF) a elektrony přenášející flavoprotein:ubichinon oxidoreduktázy (ETF:QO). Klinické fenotypy zahrnují mimo letální ne onatální formy také onemocnění s pozdním začátkem, jejichž hlavním projevem je kromě atak hypoketotické hypoglykemi e myopatický syndrom s proximálním maximem. Incidenci myopatické formy glutarové aciduri e II. typu literární zdroje ne uvádějí, nicméně díky snadnější dostupnosti moderních di agnostických metod, včetně tandemové hmotnostní spektrometri e, a rosto ucí informovanosti kliniků trvale sto upá počet nově di agnostikovaných paci entů. Kazuistika našeho paci enta, di agnostikovaného poprvé v České republice až na úrovni DNA, ilustruje jak klinický průběh onemocnění, tak možnosti di agnostiky a terapi e, a mohla by tak být inspirací pro ne urologicko u praxi.
Klíčová slova:
glutarová acidurie II. typu – mnohočetný deficit acyl-CoA dehydrogenáz – proximální myopatie – riboflavin
Úvod
Zatímco geneticky podmíněné strukturální myopatie jsou standardně předmětem diferenciálně diagnostické rozvahy, na myopatie, které jsou projevem dědičné metabolické poruchy, se někdy v klinické praxi zapomíná. Cílem tohoto článku je upozornit na metabolickou myopatii, která je diagnostikovatelná neinvazivními metodami a je i léčebně ovlivnitelná. Běžně užívaný název glutarová acidurie II. typu (GAII) je odvozen z nálezu specifických metabolitů v moči, přesnější název mnohočetný deficit acyl‑CoA dehydrogenáz (MADD) odkazuje k biochemické podstatě poruchy [1,2].
Biochemická a genetická podstata poruchy
Onemocnění je podmíněno funkčním deficitem mitochondriálních proteinů, zajišťujících přenos elektronů z dehydrogenáz, jejichž prostetická skupina je tvořena flavinadenindinukleotidem (FAD), do respiračního řetězce. Jde o spřažený systém elektrony přenášejícího flavoproteinu (ETF) a elektrony přenášející flavoprotein:ubichinon oxidoreduktázy (ETF:QO) (obr. 1). ETF je dimer tvořený α a β podjednotkou, uložený v mitochondriální matrix, kde funguje jako akceptor elektronů pro minimálně 12 dehydrogenáz (acyl‑CoA dehydrogenázy β-oxidační spirály, acyl‑CoA dehydrogenázy některých aminokyselin, protoporfyrinogen oxidáza). Je reoxidován ETF:QO, dehydrogenázou uloženou ve vnitřní mitochondriální membráně, která přes ubichinon (koenzym Q10) přenáší elektrony do respiračního řetězce. Blokáda transportního systému elektronů v mitochondrii vede k poruše řady biochemických procesů, insuficienci energetického metabolizmu a hromadění toxických metabolitů [3].
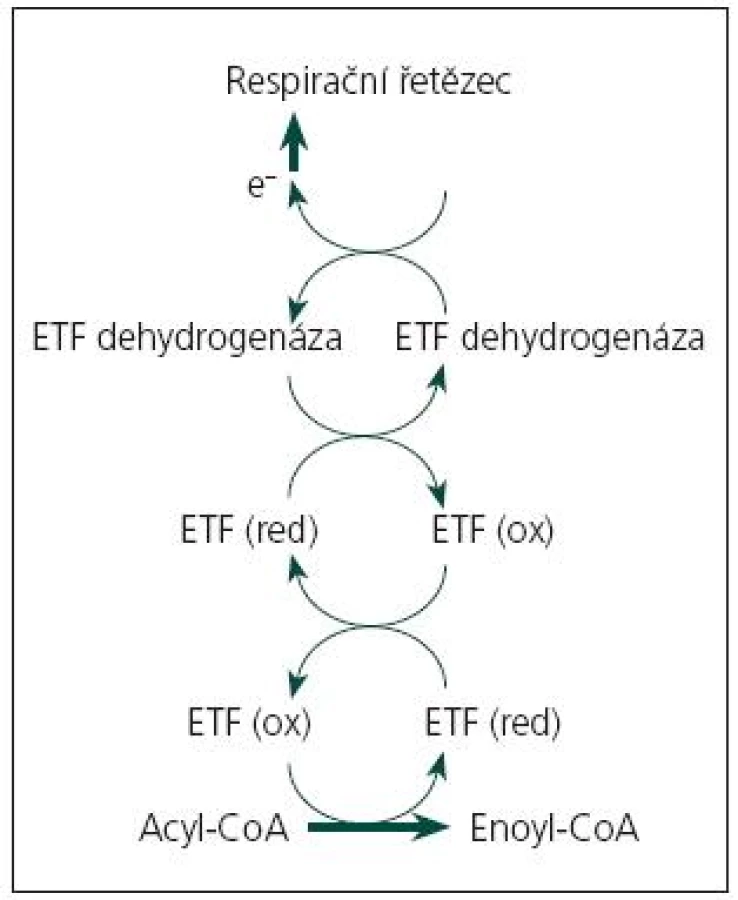
Aktuálně jsou známy tři geny, jejichž mutace jsou příčinou MADD: gen pro α podjednotku ETF (ETFA, lokus 15q23–25, MIM 608053), gen pro β podjednotku ETF (ETFB, lokus 19q13.3, MIM 130410) a gen pro ETF:ubichinon oxidoreduktázu (ETFDH, 4q32-ter, MIM 231675). Dědičnost všech dosud známých forem MADD je autozomálně recesivní [1,4,5]. V případě forem s pozdním začátkem jde často o složené heterozygoty, kdy klinický průběh poruchy u konkrétního nemocného je podmíněn mutací s mírnějšími projevy [4,5].
Někteří pacienti s late‑onset formou MADD profitují z podávání farmakologických dávek prekurzoru FAD, riboflavinu (riboflavin‑responzivní formy). Ve všech případech jde o pacienty s mutacemi v ETFDH genu. Patofyziologický mechanizmus efektu riboflavinu je stále předmětem diskuze [6].
Biochemické nálezy
Rutinní laboratorní nálezy se liší u těžkých neonatálních forem MADD a forem s pozdním začátkem. Těžké neonatální formy jsou charakterizovány výraznou metabolickou acidózou, hypoketotickou hypoglykemií, hyperlaktacidemií, elevací transamináz. S podobnými nálezy se setkáváme u late‑onset forem pouze v období dekompenzace, mimo energetickou zátěž bývají nálezy u pacientů s pozdní formou MADD normální nebo nespecifické. Mírná hepatopatie a hraniční hypoglykemie mohou uniknout pozornosti nebo mohou být spojovány s jinými noxami. Koncentrace kreatinkinázy a myoglobinu mohou být normální nebo hraničně zvýšené [7].
Rozhodující pro stanovení diagnózy jsou výsledky vyšetření zaměřeného na diagnostiku dědičných metabolických poruch. Profil organických kyselin v moči při vyšetření plynovou chromatografií s hmotnostní spektrometrií (GC/MS) charakterizovaný zvýšeným vylučováním etylmalonátu, izovalerátu, 3-hydroxyizovalerátu, glutarátu, 2-hydroxyglutarátu i dalších dikarboxylových kyselin a specifických glycinových konjugátů (např. hexanoyl-, izobutyryl-, butyryl-, izovalerylglycinu) je diagnostický. U late-onset forem MADD mohou být specifické metabolity zachyceny v moči a krvi jen v období zátěže, u riboflavin‑responzivních forem mohou na léčbě vymizet. Vyšetření suché krevní kapky tandemovou hmotnostní spektrometrií (MS/MS) prokáže specifický profil acylkarnitinů při snížené koncentraci acetylkarnitinu, tento nález zpravidla přetrvává i v období metabolické kompenzace pacientů. Hromadění acylkarnitinů může být doprovázeno deficitem volného karnitinu.
Klinický obraz a diagnostika late‑onset forem MADD
Zatímco neonatální formy nemoci mají velmi nepříznivou prognózu s letálním zakončením buď krátce po porodu, nebo v prvních měsících či letech života, průběh forem s pozdním začátkem je velmi variabilní. Jsou popsáni pacienti s prvními příznaky v dětství i v pozdní dospělosti [7], u většiny nemocných se porucha rozvíjí ve druhé či třetí životní dekádě. U některých pacientů probíhá onemocnění velmi mírně, s nevýraznou svalovou slabostí, horší tolerancí fyzické námahy, u většiny je v popředí rozvoj myopatického syndromu s proximálním maximem, oslabením šíjového svalstva, často s dekompenzací celkového stavu s vazbou na zátěž (infekce, operace, protrahovaná fyzická aktivita, hladovění). Pacienti mohou mít ataky hypoglykemie, která však v dospělosti nebývá vždy rozpoznána. Potíže jsou chronicky progresivní, rychlost a charakter progrese jsou dány jednak genotypem každého pacienta, jednak epigenetickými faktory, včetně léčby a režimu pacientů. Pečlivé zhodnocení klinického obrazu může vést velmi brzy k indikaci vyšetření dědičných metabolických poruch a stanovení diagnózy. Předpokladem je odeslání adekvátního materiálu – vzorku 5–10ml moči, vzorku krve a suché krevní kapky na krevním papírku (obr. 2) – do specializované laboratoře. U riboflavin‑responzivních forem MADD mohou při podávání riboflavinu specifické nálezy vymizet. V praxi vedou projevy pletencové myopatie s odpovídajícím EMG nálezem (myogenní vzorec) zpravidla ke svalové biopsii s variabilním nálezem, kolísajícím od normy k různě intenzivně vyjádřené myodystrofii, někdy s projevy steatózy [1,6,8,9].
Léčba
U těžkých neonatálních forem poruchy je léčba převážně symptomatická a málo úspěšná. U late‑onset forem je možnost léčebného ovlivnění podmíněna individuálním genotypem pacientů. U riboflavin‑responzivních forem je patrný pozitivní efekt farmakologických dávek riboflavinu (100–300 mg/den) rozdělených do pěti denních dávek, u pacientů s deficitem volného karnitinu je nutno podávat L-karnitin (100–200 mg/den). Vzhledem k insuficienci mitochondriální β-oxidace mastných kyselin je u všech pacientů nutná strava s vysokým obsahem sacharidů, tuky by měly být zastoupeny do 20 % energetické potřeby organizmu, podle situace je možno využít ve stravě maltodextriny nebo nevařený kukuřičný škrob. Vysloveně nevhodná je déletrvající tělesná námaha (včetně nevhodně vedené rehabilitace). V zátěži (infekce, operace, hladovění z různých důvodů) je nutné monitorování glykemie a podle potřeby zajištění intravenózního příjmu glukózy.
Genetické poradenství a prenatální diagnostika
U všech pacientů s diagnózou MADD je indikováno genetické poradenství, nejlépe na pracovišti, které má s touto problematikou zkušenosti. Vzhledem k autozomálně recesivní dědičnosti všech typů poruchy je riziko onemocnění pro sourozence 25 %, rodiče nemocných pacientů jsou obligátní heterozygoti. Prenatální diagnostika je při zájmu rodičů jednoznačně indikována u neonatálních forem onemocnění. Pokud jsou známy familiárně se vyskytující patogenní mutace, probíhá prenatální diagnostika v choriových klcích či kultivovaných amniocytech molekulárně genetickými metodami.
Kazuistika
Anamnéza a klinický obraz poruchy
42letý muž, v jehož široké rodině se u nikoho podobné potíže neobjevily. Pacient je bez perinatálních rizik, absolvoval vojenskou prezenční službu, pracoval jako tesař a zedník, do nástupu níže uvedených potíží nebyl vážněji nemocen.
Od 30 let se začal objevovat pocit únavy a svalové slabosti během pohybu doprovázený myalgiemi, zejména ve stehnech a ramenou, EMG nález svědčil pro myogenní lézi. Pro podezření na polymyozitidu byl pacient předán do péče revmatologů, léčen kortikoidy a imunosupresivy s přechodným efektem. Po dvou letech léčby svalová slabost, intolerance námahy a myalgie progredovaly, výrazněji po 37. roce věku, kdy se také poprvé objevil stav poruchy vědomí, hodnocený jako komplikovaný ortostatický kolaps.
Vyšetření materiálu z následně provedené otevřené svalové biopsie prokázalo myopatický vzorec změn s dystrofickými rysy, bez známek zánětlivého postižení. Patologická vakuolizace, změny mitochondrií nebo známky střádání patologického materiálu nebyly pozorovány na úrovni světelné, ani elektronové mikroskopie. Imunohistochemicky nebyl na 40–50 % svalových vláken prokázán dysferlin, vyšetření Western blotem prokázalo deficit kalpainu-3. Bylo vysloveno podezření na dysferlinopatii (LGMD 2B) nebo pravděpodobněji kalpainopatii (LGMD 2A), s tím, že nelze vyloučit sekundární původ těchto nálezů při jiné myodystrofii. Následná molekulárně genetická analýza mutace v genech pro dysferlin a kalpain‑3 neprokázala.
Na základě nálezu ve svalové biopsii byly postupně redukovány a vysazeny kortikoidy i imunosupresiva, pacient byl předán do péče nervosvalové poradny neurologické kliniky VFN, odkud byl odeslán materiál (krev, moč a suchá krevní kapka) k vyšetření dědičných metabolických poruch. V mezidobí při horečnatém infektu došlo k výraznému zhoršení svalové slabosti s následnou somnolencí a poruchou vědomí s křečemi, při přijetí na ARO byla prokázána těžká hypoglykemie, která se rychle upravila po intravenózní aplikaci glukózy.
V Ústavu dědičných metabolických poruch bylo z dodaného materiálu provedeno vyšetření organických kyselin v moči a séru plynovou chromatografií s hmotnostní spektrometrií a vyšetření specifických acylkarnitinů metodou tandemové hmotnostní spektrometrie v suché krevní kapce. Vyšetření prokázala přítomnost specifických metabolitů, typických pro MADD (tab. 1). Diagnóza byla podpořena patologickým nálezem při vyšetření mitochondriální β-oxidace mastných kyselin v lymfocytech. Byla zavedena adekvátní terapeutická opatření, jejichž prostřednictvím se podařilo stabilizovat klinický stav pacienta.

Objektivní neurologický nález při stanovení diagnózy
Bez známek kognitivního deficitu, dyspnoe a hyperhidróza v zátěži, difuzní palpační bolestivost svalstva, bez svalových kontraktur. Normální nález na hlavových nervech. Oslabení proximálních svalových skupin, zejména proti odporu, ochablá břišní stěna. Oslabení šíjového svalstva, aktivní předklon hlavy vleže na zádech je však možný. Kvadruhyporeflexie, pokles končetin v polohách na výdrž, zhoršování taxe při opakování zkoušek, negativní iritační pyramidové jevy. Pacient se posadí a postaví namáhavě jen s pomocí paží, chůze v hyperlordóze, kolébavá, ujde obtížně několik metrů v místnosti. Chůzi po špičkách pacient nezvládne, na patách je schopen s obtížemi provést několik kroků.
Terapie a současný klinický průběh poruchy
Pacient a ošetřující lékaři byli informováni o požadavcích na pohybový a dietní režim (viz kapitola Léčba). Vzhledem k možnosti riboflavin‑responzivní formy byl u pacienta od počátku nasazen riboflavin v dávce 60 a posléze 100 mg denně (rozděleno do pěti denních dávek). Jsou monitorovány koncentrace karnitinu, přechodný deficit byl upraven, proto není pacient aktuálně suplementován (tab. 1).
Při zavedených opatřeních pletencová myopatie neprogreduje. Porucha vědomí s křečemi se neopakovala. Pacient je schopen při dodržování vhodného tempa ujít i několik stovek metrů, bez pomoci francouzských holí, plně zvládá sebeobsluhu. Hlavním problémem při aktuálním zlepšení mobility jsou sekundární změny na skeletu kyčlí, pravděpodobně s uplatněním dlouhodobé kortikoterapie v minulosti. Kardiologický nález včetně echokardiografie je normální.
Monitorování specifických metabolitů při zavedeném režimu a terapii riboflavinem prokázalo jejich vymizení v moči, přetrvává přítomnost specifických acylkarnitinů při vyšetření suché krevní kapky tandemovou hmotnostní spektrometrií (tab. 1).
Molekulárně genetické vyšetření
Ve spolupráci s výzkumnou molekulárně genetickou laboratoří University of Aarhus, specializující se na genotypizaci MADD, byla provedena mutační analýza v ETFA, ETFB a ETFDH genu v genomové DNA pacienta a jeho rodičů, která potvrdila, že pacient je složený heterozygot pro dvě různé patogenní alely podmíněné bodovými mutacemi v ETFDH genu (gen pro elektrony přenášející flavoprotein:ubichinon oxidoreduktázu – ETF:QO). Mutace c.652G > A (p.Asp218Asn) v exonu 6 ETFDH genu vede v homozygotní formě k těžkému průběhu onemocnění, mutace c.1366C > T (p.Pro456Ser) v exonu 11 ETFDH genu je hodnocena jako mírná. Mutace vedoucí k záměně prolinu ve výše uvedené pozici za jinou aminokyselinu jsou popisovány jako nejčastější u pacientů s riboflavin‑responzivní formou choroby [4,5].
Diskuze
Význam otevřené svalové biopsie s komplexním vyšetřením materiálu zkušeným specialistou je pro diagnostiku progresivních myopatií všeobecně považován za zásadní [1,8,9]. Přítomnost specifických klinických příznaků by však měla vést klinika i k úvaze o možnosti metabolicky podmíněné myopatie s následným odesláním materiálu do specializované laboratoře [1,2,7].
Materiálem, jehož vyšetření má největší přínos pro diagnostiku poruch β-oxidace mastných kyselin, včetně MADD, je suchá krevní kapka (obr. 2). I při normálních nálezech v tělesných tekutinách (moč, sérum), jak tomu bývá u nemocných s MADD v kompenzovaném stavu, lze v erytrocytech ze suché krevní kapky obvykle prokázat diagnostické metabolity – specifické acylkarnitiny, které se v erytrocytech hromadí a jen minimálně přecházejí do séra. Jejich koncentrace proto dosahují vůči séru zhruba trojnásobných hodnot a jsou do určité míry stabilní [1,2,4–7]. Nezanedbatelnou výhodou tohoto materiálu je rovněž nekomplikovaná doprava poštou.
Autozomálně recesivní pletencové muskulární dystrofie (LGMD2) patří k relativně častějším hereditárním myopatiím dospělého věku [8,9]. Jde o heterogenní skupinu poruch spojených většinou s deficitem strukturálních svalových proteinů, u deficitu kalpainu-3 (LGMD2A) však jde o deficit neutrální svalové proteázy, účastnící se procesu nekrózy poškozených svalových buněk, a u deficitu dysferlinu (LGMD2B) o defekt reparačního proteinu membrán svalových buněk, tedy o proteiny funkční [8,9]. Patofyziologie obou proteinů není dosud zcela objasněna, dosavadní zkušenosti nevylučují i sekundární změny v jejich expresi v souvislosti s jiným myodystrofickým procesem a/nebo epigenetickými vlivy. U pacienta s riboflavin respozivní formou MADD se nabízí otázka, zda by došlo k regresu imunohistochemického nálezu deficitu dysferlinu na léčbě riboflavinem. Odpověď by mohla přinést kontrolní svalová biopsie, jejíž indikace je však při zlepšeném klinickém stavu pacienta problematická.
Responzibilitou na riboflavin u pacientů s MADD se zabývala v posledních letech Olsen se spolupracovníky [5]. U všech riboflavin‑responzivních pacientů byly prokázány mutace v ETFDH genu. Mechanizmus efektu riboflavinu není zatím jednoznačně znám, předpokládá se, že zvýšená nabídka prekurzoru (riboflavinu) vede k vyšším koncentracím FAD, jenž je jako kofaktor nezbytný pro zajištění adekvátní konformace a stability ETF:QO, působí tedy jako chaperon. Některé mutace v ETFDH genu vedou ke snížení afinity apoenzymu k FAD, v těchto případech může zvýšení koncentrace kofaktoru (FAD) zvýšit tvorbu funkčního holoenzymu.
Skutečnost, že některé geneticky rozdílné poruchy se fenotypicky překrývají, zdůvodňuje, proč je důležitá komplexní diagnostika myopatických syndromů. Příkladem mohou být někteří pacienti s deficitem koenzymu Q10, u nichž byl prokázán primární defekt ETF:QO, pozitivně reagující na riboflavin [6]. Diferenciálně diagnosticky je nutno podle současných poznatků pomýšlet na riboflavin‑responzivní formu MADD i u některých pacientů s deficitem komplexu I, II a IV respiračního řetězce [5,6].
Závěr
Late‑onset forma mnohočetného deficitu acyl‑CoA dehydrogenáz je nedostatečně diagnostikované, geneticky podmíněné, klinicky heterogenně probíhající onemocnění, jehož významným projevem je různě rychle progredující pletencová myopatie. Diagnostický proces u myopatických syndromů s proximálním maximem by proto měl mimo standardně provedenou a zhodnocenou svalovou biopsii zahrnovat i vyšetření moči, krve a suché krevní kapky ve specializované laboratoři zabývající se diagnostikou dědičných metabolických poruch.
Práce byla podpořena výzkumným záměrem MZOVFN2005 MZ ČR a VZ FNM 00064203.
MUDr. Helena Jahnová
Ústav dědičných metabolických poruch
VFN a 1. LF UK v Praze
Ke Karlovu 2
128 08 Praha 2
e-mail: helena.jahnova@vfn.cz
Sources
1. Frerman FE, Go odman SI. Defects of electron transfer flavoprotein and electron transfer flavoprotein‑ubiquinone oxidoreductase: glutaric acidemi a type II. In: Scriver CR, Be a udet AL, Sly WL, Valle D (eds). The metabolic and molecular bases of inherited dise ase. New York: McGraw- Hill 2001: 2357– 2365.
2. Duran M. Disorders of Mitochondri al Fatty Acid Oxidati on and Ketone Body Handling. In: Bla u N, Duran M, Blaskovics ME, Gibson KM (eds). Physici an’s guide to the laboratory di agnosis of metabolic dise ases. 2nd ed. Heidelberg: Springer- Verlag 2003: 309– 334.
3. Depeint W, Bruce R, Shangari N, Mehta R, O’Bri en PJ. Mitochondri al functi on and the B- vitamin family on mitochondri al energy metabolism. Chem Bi ol Interact 2006; 163(1– 2): 94– 112.
4. Olsen RK, Andresen B, Christensen E, Bross P, Skovby F, Gregersen N. Cle ar relati onship between ETF/ ETFDH genotype and phenotype in pati ents with multiple acyl- CoA dehydrogenati on defici ency. Hum Mutat 2003; 22(1): 12– 23.
5. Olsen RK, Olpin SE, Andresen BS, Mi edzybrodzka ZH, Po urfarzam M, Merinero B et al. ETFDH mutati ons as a major ca use of riboflavin‑responsive multiple acyl‑ CoA dehydrogenati on defici ency. Brain 2007; 130(8): 2045– 2054.
6. Gempel K, Topaloglu H, Talim B, Schneiderat P, Schosser BGH, Hans VH et al. The myopathic form of coenzyme Q10 defici ency is ca used by mutati ons in the electron- transferring- flavoprotein dehydrogenase (ETFDH) gene. Brain 2007; 130(8): 2037– 2044.
7. Köppel S, Gottschalk J, Hoffmann GF, Waterham HR, Blobel H, Kölker S. Late‑onset multiple acyl‑ CoA dehydrogenase defici ency: a frequently missed di agnosis? Ne urology 2006; 67(8): 1519.
8. Bednařík J et al (eds). Nemoci kosterního svalstva. Praha: Triton: 2001: 136– 142.
9. Angelini C, Nardetto L, Fanin M, Nascini AC, Tasca E.Heterogeno us pathogenesis of LGMD2: consequences for therapy. Basic Appli ed Myology 2007; 17(3– 4): 173– 179.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2009 Issue 3
Most read in this issue
- Autologo us Hematopo i etic Stem Cells Transplantati on and its Current Role in Multiple Sclerosis Tre atment
- Migraine
- Adult Form of Glutaric Aciduri a Type II – Under di agnosed Ca useof Proximal Myopathy – a Case Report
- Transfer of the Tibial Posteri or Muscle Tendon – Effici ent Soluti on to Perone al Muscular Paresis