
Leberova hereditární optická neuropatie s oligoklonálními pásy v likvoru považovaná za roztro ušeno u sklerózu – kazuistika
Leber’s Hereditary Optic Neuropathy with Oligoclonal Bandsin the Serum Regarded as Multiple Sclerosis – a Case Report
Visi on deteri orated in an 18- ye ar-old man witho ut peri ocular pain during one week in the left eye first and in the right eye two months later. Ne urological findings were otherwise normal. Magnetic resonance imaging (MRI) of the brain was normal and there were high signals within optic nerves and chi asma. Oligoclonal IgG bands were repe atedly detected in the cerebrospinal fluid. As a result, the disorder was considered to be multiple sclerosis. The visi on failed to improve during one- ye ar follow‑up and no other impairment appe ared. The results of a uxili ary investigati ons were not in agreement with the atypical clinical co urse of the dise ase and normal MRI of the brain. We considered other di agnoses due to this discrepancy. Anti‑aqu aporin antibodi es did not support ne uromyelitis optica (Devic dise ase). Finally, the mitochondri al DNA mutati on confirmed Leber hereditary optic ne uropathy (LHON). The correct di agnosis was achi eved after bro ad interdisciplinary co operati on including foreign laboratory. This case illustrates the misle ading role of oligoclonal bands in cerebrospinal fluid and sho uld incre ase general knowledge about LHON in physici ans.
Key words:
multiple sclerosis – Leber hereditary optic ne uropathy – oligoclonal IgG bands – cerebrospinal fluid
Authors:
L. Čechová 1; A. Bartoš 1,2; J. Bartošová 3; D. Doležil 1
Authors‘ workplace:
Univerzita Karlova 3. LF a FN Královské Vinohrady, Neurologická klinika, Praha, 2Psychiatrické centrum Praha, 3Univerzita Karlova 3. LF a FN Královské Vinohrady, Oftalmologická klinika, Praha
1
Published in:
Cesk Slov Neurol N 2009; 72/105(2): 155-158
Category:
Case Report
Overview
U 18letého mladíka se během jednoho týdne významně zhoršilo vidění nejdříve levým a po dvo u měsících i pravým okem bez peri okulární bolesti. Ostatní neurologický nález byl v normě. Na magnetické rezonanci (MR) mozku byl normální obraz, v optických nervech a chiazmatu se nalezla tečkovitá hypersignální ložiska. V likvoru byly opakovaně přítomny oligoklonální pásy IgG. Proto bylo onemocnění považováno za roztro ušeno u sklerózu. Během jednoho roku sledování se zrak nezlepšil a neobjevilo se žádné další postižení. Výsledky pomocných vyšetření byly v rozporu s atypickým klinickým průběhem a normálním MR obrazem. Tento neso ulad nás vedl k podezření na jiné di agnózy. Antiakvaporinové protilátky nepodpořily možnost Devicovy optické neuromyelitidy. Nakonec se prokázala mutace v mitochondri ální DNA svědčící pro Leberovu hereditární opticko u neuropatii (LHON). Ke správnému určení vzácné diagnózy u složitého pacienta přispěla široká mezioborová spolupráce včetně zahraniční laboratoře. Případ dokládá zavádějící úlohu oligoklonálních pásů v likvoru a má směřovat k většímu povědomí o LHON v odborné veřejnosti.
Klíčová slova:
roztro ušená skleróza – Leberova hereditární optická ne uropati e – oligoklonální pásy IgG – mozkomíšní mok
Úvod
Porucha nebo až ztráta zraku je u mladého jedince alarmujícím příznakem a bývá výzvou k mezioborové spolupráci mezi neurologem a oftalmologem. Zhoršené vidění má řadu příčin, mezi něž patří onemocnění zrakového nervu různé etiologie. Výpadky v zorném poli jsou často způsobeny záněty při demyelinizačních chorobách (idiopatická optická neuritida – ON; ON při roztroušené skleróze, Devicova optická neuromyelitida – NMO, akutní diseminovaná encefalomyelitida). Naopak vzácným postižením je Leberova hereditární optická neuropatie (LHON) [1].
LHON je způsobena bodovými mutacemi v mitochondriální DNA (mtDNA). Patří mezi nejčastější mitochondriální onemocnění. Prevalence není přesně známá, odhaduje se zhruba 1 : 25 000 [2]. Onemocnění popsal poprvé německý oftalmolog Theodor Leber již v roce 1871. Trvalo více než 100 let, než Wallace et al odhalili mutaci G11778A v mtDNA [3]. Od té doby výrazně pokročily poznatky o této nemoci. V 95 % případů je přítomna jedna z mutací G3460A, G11778A a T14484C [2–4]. Dědičnost je maternální, nemocné matky přenesou mutaci na všechny děti. Žádný z potomků nemocných mužů nebude postižen. Nemoc se vyznačuje inkompletní penetrancí, mnohem častěji bývají postiženi muži [2]. Je vysoce pravděpodobné (i když ne dosud potvrzeno), že penetranci ovlivňují také faktory zevního i vnitřního prostředí jako kouření, alkohol, nutriční deficit, stres a akutní onemocnění [4].
Porucha zraku se typicky objevuje mezi 15. až 35. rokem věku. Postižení začíná bezbolestně nejdříve na jednom oku. Za několik týdnů či měsíců (průměrně osm týdnů) se rozvine i na oku druhém, méně často na obou očích současně. Charakteristický výpadek zorného pole u LHON je centrocekální skotom. Degenerací retinálních nervových vláken postupně dochází k atrofii zrakového nervu. Intenzita postižení zraku kolísá od nerozeznávání světla až k normálnímu vizu. U většiny pacientů je zrak horší než 6/60. Na očním pozadí může být v akutní fázi přítomna triáda: pseudoedém papily, cirkumpapilární telengiektatická mikroangiopatie, absence prosakování barviva z papily a cév při fluorescenční angiografii (FAG). U některých pacientů se však ani v akutní fázi nemusí tato triáda vyskytnout, proto její nepřítomnost LHON nevylučuje [4]. Magnetická rezonance (MR) mozku bývá většinou normální, někdy jsou patrné hypersignální změny uvnitř zrakového nervu bez postkontrastního sycení. Vizuální evokované potenciály (VEP) potvrdí postižení zrakového nervu [2].
V některých případech (zřetelně častěji u žen s mutací G11778A) se rozvinou klinické příznaky a MR obraz nerozeznatelné od roztroušené sklerózy (RS), tzv. LHON-MS syndrom [2,5,6].
Není dostupná žádná kauzální léčba, podává se pouze podpůrná vitaminoterapie. V budoucnu by mohla znamenat šanci genová terapie. Pacientům a jejich rodinným příslušníkům lze nabídnout genetické poradenství [2,5].
Na příkladu mladého pacienta chceme ilustrovat svízele při diagnostice LHON, o níž bylo dosud v českém písemnictví referováno minimálně [7]. Pravděpodobně malé povědomí o vzácném onemocnění a přítomnost oligoklonálních pásů (OCB) IgG v mozkomíšním moku vedly zpočátku k záměně za RS.
Kazuistika
U 18letého mladíka se zhoršilo vidění nejdříve levým a po dvou měsících i pravým okem. Těžká porucha zraku se rozvinula vždy bez bolesti během jednoho týdne. Dvojité vidění, parestezie, poruchu hybnosti, ani žádné další obtíže nepozoroval. V osobní anamnéze udával častější záněty středouší, operaci nesestouplého varlete, excizi névu na břiše. Úrazy negoval. Klíšťata míval opakovaně každý rok. Pracoval jako dělník s plasty, při tavení vdechoval chemické výpary, kouřil 15 cigaret denně. V rodině je známo několik případů těžké poruchy zraku, a to u matčina strýce, u dvou synů matčiny tety a u matčiny sestry. Poslední jmenovaná je z neznámých důvodů na invalidním vozíku a je údajně léčena pro RS. Bližší informace o jejím zdravotním stavu se i přes naši snahu nepodařilo zjistit. Jiní rodinní příslušníci anamnesticky neurologické obtíže nemají.
Objektivně byl přítomen normální neurologický nález při oboustranné těžké poruše vizu. Pacient vyhledal odbornou pomoc až po zhoršení zraku druhého oka. Časná fáze onemocnění také nebyla důkladně vyšetřena, ani FAG nebyla provedena. Elektroretinografie a klinický nález po půlroce od vzniku obtíží nesvědčily pro difuzní dystrofické onemocnění sítnice. VEP prokázaly oboustrannou abnormitu odpovídající prechiazmatické lézi s výraznou amplitudovou depresí a prodloužením latencí. Na MR mozku byla popsána pouze diskrétní tečkovitá hypersignální ložiska v optických nervech a chiazmatu jen na sekvencích FSTIR bez postkontrastního sycení (obr. 1). Jinak byl MR obraz mozku bez demyelinizačních ložisek či jiných patologií. V likvoru byly přítomny čtyři oligoklonální pásy v alkalické oblasti a pozitivní antiboreliové protilátky IgG stanovené ELISA, ale při vyšetření western blotem nebyla přítomnost antiboreliových protilátek potvrzena. Zvažovala se boreliová optikopatie či spíše počínající demyelinizační onemocnění. Nakonec byl stav na spádovém neurologickém pracovišti uzavřen jako RS a byla zahájena kombinovaná imunosupresivní léčba.
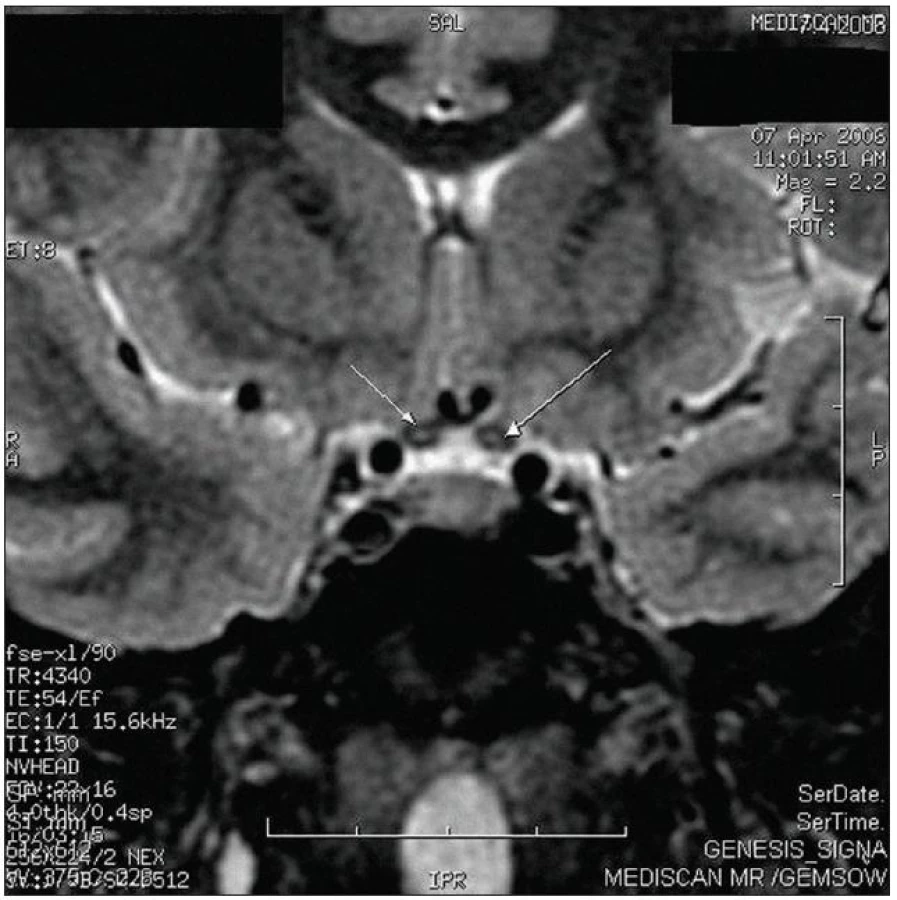
Po jednom roce trvání obtíží se pacient objevil v naší neuroimunologické poradně Neurologické kliniky 3. LF UK a FNKV. Zrak se nezlepšil a nevzniklo žádné nové postižení. Pro atypický klinický průběh a normální MR mozku jsme uvažovali o dalších příčinách onemocnění. Po domluvě s pacientem jsme zopakovali vyšetření likvoru. Základní biochemický nález byl normální a počet mononukleárů byl 48/3. Antiboreliové protilátky byly již negativní v likvoru i v séru. Přetrvávaly oligoklonální pásy IgG v alkalické oblasti v mozkomíšním moku, jejichž počet se zvýšil ze 4 na 13 (avšak stanovení proběhlo na jiném pracovišti, obr. 2).

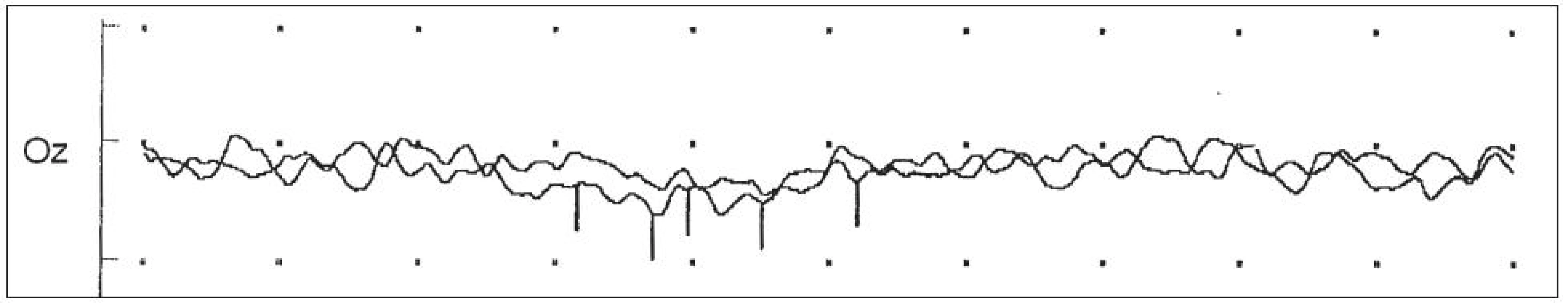
Na kontrolním vyšetření VEP po roce byla přítomna nehodnotitelná křivka z obou očí (obr. 3). Doplnili jsme kmenové, motorické i somatosenzorické evokované potenciály s normálními nálezy. Při imunologickém vyšetření nebyla detekována významná abnormita. Protilátky proti akvaporinu‑4 v séru nemocného nebyly přítomny. Na kontrolní MR mozku a krční míchy po 1 i 2,5 letech byl fyziologický nález, již bez tečkovitého zvýšení signálu v optických nervech. Při očním vyšetření byl nález stacionární. Vizus obou očí byl 1/60. Na předním očním segmentu byl normální nález, na fundu bilaterálně tilted disk s temporálním nablednutím, jinak bez abnormit. Na perimetru (120 bodů) byly výpady centrální i v periferii difuzně oboustranně.
I přes přítomnost OCB v moku jsme vzhledem k izolované poruše vidění s normálním neurologickým nálezem a normální MR mozku zvažovali LHON. Přítomnost mutace G3460A v mtDNA v krvi a ve vlasových folikulech LHON potvrdila. Imunosupresivní léčbu jsme nahradili vitaminoterapií. Rodině byla vysvětlena povaha onemocnění a byly provedeny genetické analýzy řady členů rodiny. Mutace byla nalezena u pacientovy matky, sestry matky a jejího syna i dcery. U žádného z nich se onemocnění dosud neprojevilo. U bratrance nemocného se pro nespolupráci nepodařilo doplnit plánovanou MR mozku ani neurologické vyšetření. U sestry matky s postižením zraku údajně léčené jako RS nebylo možné ověřit diagnózu vzhledem k jejímu odmítavému stanovisku k dalšímu vyšetřování.
Diskuze
Náš případ dokládá, že u pacienta s LHON se mohou vyskytnout OCB v mozkomíšním moku, aniž by v současné době trpěl RS. OCB nacházíme v 95 % případů RS a jejich přítomnost při první atace neurologických obtíží předvídá pravděpodobný rozvoj klinicky jisté RS [8]. Vysoká senzitivita OCB v moku pro RS a malé povědomí o vzácné LHON proto zpočátku vedly k interpretaci očních obtíží jako manifestace RS. Diagnóza RS byla také podpořena dvěma oddělenými epizodami poruchy vidění. Avšak přetrvávající selektivní oboustranné postižení zrakových nervů a normální obraz MR mozku připouštěly i jinou diagnózu.
Antiboreliové protilátky se při opakovaném vyšetření likvoru již neprokázaly. Pomýšleli jsme také na Devicovu optickou neuromyelitidu. Pacient však neměl známky myelopatie ani pozitivní antiakvaporinové protilátky v séru, které se u NMO často vyskytují [8]. Z těchto důvodů jsme u pacienta nakonec neprovedli MR míchy. Protože postižení zůstávalo klinicky i při vyšetření pomocnými metodami ohraničeno jen na zrakové nervy, uvažovali jsme o oční afekci. Pro autozomálně dominantní atrofii optiku (Kjerovu nemoc) nenacházíme typickou rodinnou anamnézu. Nakonec se geneticky potvrdilo podezření na LHON.
Výskyt OCB u LHON je v literatuře popisován vzácně. Nalezli jsme jediný případ 15letého německého chlapce s LHON a OCB v moku a přitom bez dalšího neurologického postižení [9]. Častěji OCB nacházíme u pacientek s tzv. LHON-MS syndromem. Jedná se o kombinaci LHON s MR obrazem a klinickými příznaky nerozeznatelnými od RS. Vyskytuje se téměř výhradně u žen s mutací G11778A. Výskyt tohoto MS‑like onemocnění u LHON je častější, než by odpovídalo náhodné koincidenci. LHON je pokládána za rizikový faktor pro vznik RS [2,5,6]. Úlohu zde má pravděpodobně autoimunita, v porovnání s kontrolami byla nalezena u LHON vyšší hladina protilátek proti tubulinu optického nervu [2].
V našem případě byla přítomnost OCB zavádějící a způsobila záměnu za RS. Přítomnost OCB nám nepřipadala zcela příhodná k celkovému klinickému kontextu, ale byla prokázána opakovaným vyšetřením likvoru s časovým odstupem, a to dokonce na dvou různých pracovištích. Důvod autochtonní produkce IgG u našeho pacienta není jasný. Může to být následek nějaké latentní neuroinfekce v rámci běžné virózy v minulosti, při nichž OCB mohou přetrvávat roky, aniž jsou spojeny s nějakými neurologickými příznaky [10,11]. Otázkou je, zda jsou oligoklonální pásy u LHON častějším jevem, nebo zda jsou nepříznivým prognostickým znamením pro vznik LHON-MS syndromu. Náš pacient má izolovanou poruchu vidění již tři roky a zatím se u něj žádné neurologické postižení neobjevilo. Uvidíme v budoucnu, zda se u něj RS přeci jen rozvine či nikoli.
LHON je vhodné zařadit do diagnostických rozvah u všech jedinců s poměrně náhle vzniklou poruchou vizu. Podezření by mělo vzbudit zejména oboustranné postižení bez periokulární bolesti u mladého muže. Při diagnostických rozpacích lze v akutní fázi podpořit možnost LHON vyšetřením FAG. Důležité je zaměřit se také na rodinnou anamnézu. Naše podezření zesílí, pokud nejsme schopni potvrdit RS pomocnými metodami, a to ani v časovém odstupu. Hlavní význam odlišení spočívá v tom, že nemocní s domnělou RS jsou zbytečně vystavováni imunosupresivům. Tato léčba není u LHON účinná a navíc nemocného zatěžuje potenciálními vedlejšími účinky.
Práce byla podpořena výzkumným záměrem Patofyziologie neuropsychiatrických onemocnění MSM0021620816.Děkujeme prof. Angele Vincent, MD, PhD z Oxfordské neuroimunologické laboratoře za vyšetření antiakvaporinových protilátek v séru nemocného.
MUDr. Linda Čechová
UK 3. LF a FNKV, Praha
Neurologická klinika
Šrobárova 50
100 34 Praha 10
e‑mail:
lindacechova@centrum.cz
Sources
1. Vaněk I, Bartošová J, Bartoš A. Ne uro oftalmologi e. In: Kuchynka P et al. Oční lékařství. Praha: Grada 2007: 501– 554.
2. Man PY, Turnbull DM, Chinnery PF. Leber hereditary optic ne uropathy. J Med Genet 2002; 39(3): 162– 169.
3. Oguchi Y. Past, present, and future in Leber’s hereditary optic ne uropathy – a revi ew. Jpn J Ophthalmol 2002; 46(3): 352– 353.
4. Newman NJ. Hereditary optic ne uropathi es: from the mitochondri a to the optic nerve. Am J Ophthalmol 2005; 140(3): 517– 523.
5. Teive HA, Tro i ano AR, Raskin S, Werneck LC. Leber’s hereditary optic ne uropathy – case report and literature revi ew. Sao Pa ulo Med J 2004; 122(6): 276– 279.
6. Vanopdenbosch L, Dubo is B, D’Ho oghe MB, Meire F,Carton H. Mitochondri al mutati ons of Leber’s hereditary optic ne uropathy: a risk factor for multiple sclerosis. J Ne urol 2000; 247(7): 535– 543.
7. Konrádová V, Zeman J, Stratilová L, Heřmanská, Všetička I, Mišovicová N et al. Leberova hereditární ne uropati e optiku (LHON). Čas Lék čes 1999; 138(18): 565– 568.
8. Bartoš A. Klinické využití protilátek u roztro ušené sklerózy. Cesk Slov Ne urol N 2008; 71/ 104(1): 26– 33.
9. Jung S, Fassbender K. Oligoklonale IgG- Banden im Liquor bei einem Jungen mit Leber’scher hereditärer Opticusatrophi e ohne Anhalt für Multiple Sklerose – Risikofaktor für eine LHON- MS oder „häufigeres Phänomen“? Akt Ne urol 2006; 33: 1055.
10. Bartos A, Pitha J. Opsoclonus- myoclonus- dysequilibri um syndrome: cytological and immunological dynamics in the seri al cerebrospinal fluid in two pati ents. J Ne urol 2003; 250(12): 1420– 1425.
11. Reiber H, Peter JB. Cerebrospinal fluid analysis: dise ase‑related data patterns and evalu ati on programs. J Ne urol Sci 2001; 184(2): 101– 122.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2009 Issue 2
Most read in this issue
- Krční myelopatie – diagnostický problém
- Neurodegenerativní demence
- Maligní tumor z pochvy periferního nervu – dvě kazuistiky
- Radi ofrekvenční terapi e facetových bolestí bederní páteře