
Syndrom Smithové-Magenisové: kazuistika
Smith-Magenis syndrome: a case report
Smith-Magenis syndrome is a rare disease caused by haploinsufficiency of the RAI 1 (retinoic acid-induced) gene associated with the deletion of chromosome 17p11.2 and/or its mutation. The clinical picture comprises mental retardation, behavioural disorders with marked proneness to self-injurious outbursts, and disordered sleep. Sleep disorders characterised by the overall sleep time shortening, premature awakening and increased daytime sleepiness are very typical features for the syndrome. The cause lies in inverted circadian secretion of melatonin culminating in daytime hours. Patients display characteristic craniofacial dysmorphia, and often multisystem involvement. A case of a female patient is presented, with a typical clinical picture, sleep aberrations and genetic symptoms.
Key words:
Smith-Magenis syndrome – circadian rhythm disorder – melatonin
:
I. Příhodová 1; D. Kemlink 1; K. Veselá 2; R. Mihalová 2; S. Nevšímalová 1
:
Neurologická klinika 1. LF UK a VFN v Praze
1; Ústav biologie a lékařské genetiky 1. LF UK a VFN v Praze
2
:
Cesk Slov Neurol N 2008; 71/104(2): 223-227
:
Case Report
Syndrom Smithové-Magenisové je vzácné onemocnění podmíněné haploinsuficiencí RAI 1 (retinoic acid-induced) genu spojenou s delecí chromozomu 17p11,2 nebo s jeho mutací. V klinickém obraze se kombinuje mentální retardace, porucha chování s nápadným sebepoškozováním a porucha spánku. Porucha spánku se zkrácením celkové doby spánku, předčasným probouzením a zvýšenou denní spavostí je pro tento syndrom velmi specifická. Její příčinou je obrácená cirkadiánní sekrece melatoninu s maximem v denních hodinách. U pacientů je patrná charakteristická kraniofaciální dysmorfie a bývá přítomno další multisystémové postižení. Autoři popisují pacientku s typickým klinickým obrazem onemocnění, odchylkami ve vyšetření spánku a genetickým nálezem.
Klíčová slova:
syndrom Smithové-Magenisové – porucha cirkadiánní rytmicity – melatonin
Úvod
Syndrom Smithové-Magenisové je vzácné, geneticky podmíněné onemocnění poprvé popsané v roce 1982 [1]. Je charakterizováno typickými klinickými projevy, kraniofaciální dysmorfií a dalším multisystémovým postižením. Ve většině případů (v 70–90 %) je spojeno s intersticiální delecí krátkého raménka chromozomu 17 (17p11.2) [2,3], v menším procentu s mutací RAI 1 (retinoic acid-induced) genu, který leží v kritické oblasti uvedeného úseku [4]. Frekvence výskytu je odhadována 1 na 25 000 živě narozených dětí [2]. Klinické příznaky zahrnují různě závažnou mentální retardaci, výrazně opožděný rozvoj řeči, poruchy chování se sebepoškozováním a poruchu spánku ve smyslu narušené cirkadiánní rytmicity.
Kazuistika
13letá pacientka byla přijata na vyšetření pro nadměrnou denní spavost až imperativního rázu a pro nekvalitní noční spánek s předčasným ranním probouzením. Dívka se narodila ze 4. fyziologického těhotenství, porod byl ve 40. týdnu, indukovaný, záhlavím, porodní hmotnost měla 3 120 g Nebyla kříšena, pro hyperbilirubinemii byla indikována fototerapie. Pro kraniofaciální dysmorfii proběhlo již v novorozeneckém věku vyšetření karyotypu k vyloučení Downova syndromu a dále pak pro přetrvávající hypotonický syndrom také vyšetření se zaměřením na Praderové-Williho syndrom. Obě vyšetření byla negativní. Dívka měla mírně opožděný psychomotorický vývoj (samostatná chůze od 18. měsíce), později vázl rozvoj řeči a bylo vysloveno podezření na vývojovou dysfazii. Od 3 let byla opakovaně pro vývojové opoždění vyšetřována. CT mozku zachytilo mírnou ventrikulomegalii, EEG, endokrinologické vyšetření a screening vrozených metabolických vad byly bez odchylek, stejně tak i vyšetření sluchu Pro divergentní strabizmus, myopii a astigmatizmus byla v péči očního lékaře. Ortopedicky byla sledována pro plochonoží a stáčení pravé nohy navnitř. Vzhledem k lehké mentální retardaci chodila do speciální školy. Měla výrazné poruchy chování se záchvaty vzteku a tendencemi k sebepoškozování, bila hlavou o zeď, okusovala si nehty, kousala se do rukou. Z intenzity těchto projevů bylo zřejmé, že měla sníženou reakci na bolest. Projevovala se tendence ke stereotypním pohybům (tleskání rukama, pohyby hlavou), velmi špatně si zvykala na změny denního režimu. Trpěla hyperaktivitou, nesoustředěností a častými výkyvy nálady – záchvaty agresivity se střídaly s nadměrnou emoční příchylností.
Odmalička měla abnormální spánkový rytmus, více spala během dne a v noci se častěji budila. Zhoršení nastalo v posledním roce. Usínala bez potíží kolem 20. hodiny, ale opakovaně se během noci budila. Noční spánek byl zkrácený, s předčasným ranním probuzením (mezi 3.–4. hodinou), někdy ještě s opětovným usnutím. Během dne byla zvýšeně ospalá, pravidelně usínala odpoledne na 30–60 min. Kromě toho se objevovalo imperativní usínání při jídle či rozhovoru, někdy i při chůzi. V noci dívka chrápala, zástavy dechu ani jiné problémy ve spánku neměla, kromě sporadické noční enurézy.
Při objektivním vyšetření bylo patrné eretické chování, stereotypní pohyby, omezená slovní zásoba, hrubý hlas a dysartrie, divergentní strabizmus levého oka, pedes planovalgi. Dívka chodila o širší bázi a v mírném předklonu. Byla zřejmá kraniofaciální dysmorfie – čtverhranný obličej s hrubými rysy, srostlé obočí, hypertelorizmus, krátké filtrum, dysplastické ušní boltce (obr. 1). Na končetinách byla brachydaktylie a na dolních končetinách ještě syndaktylie 2. a 3. prstu (obr. 2).
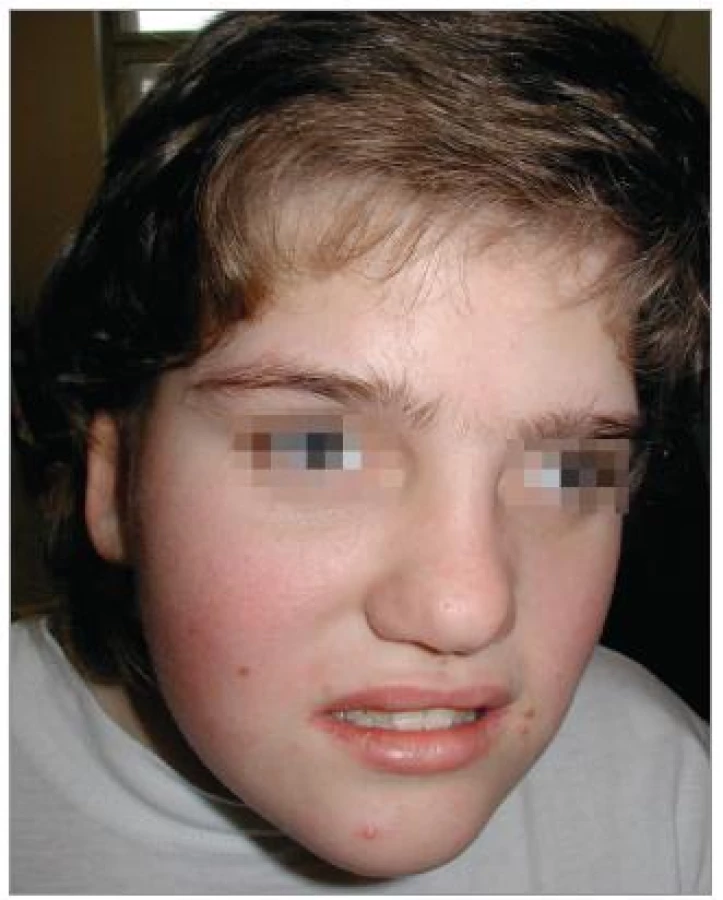
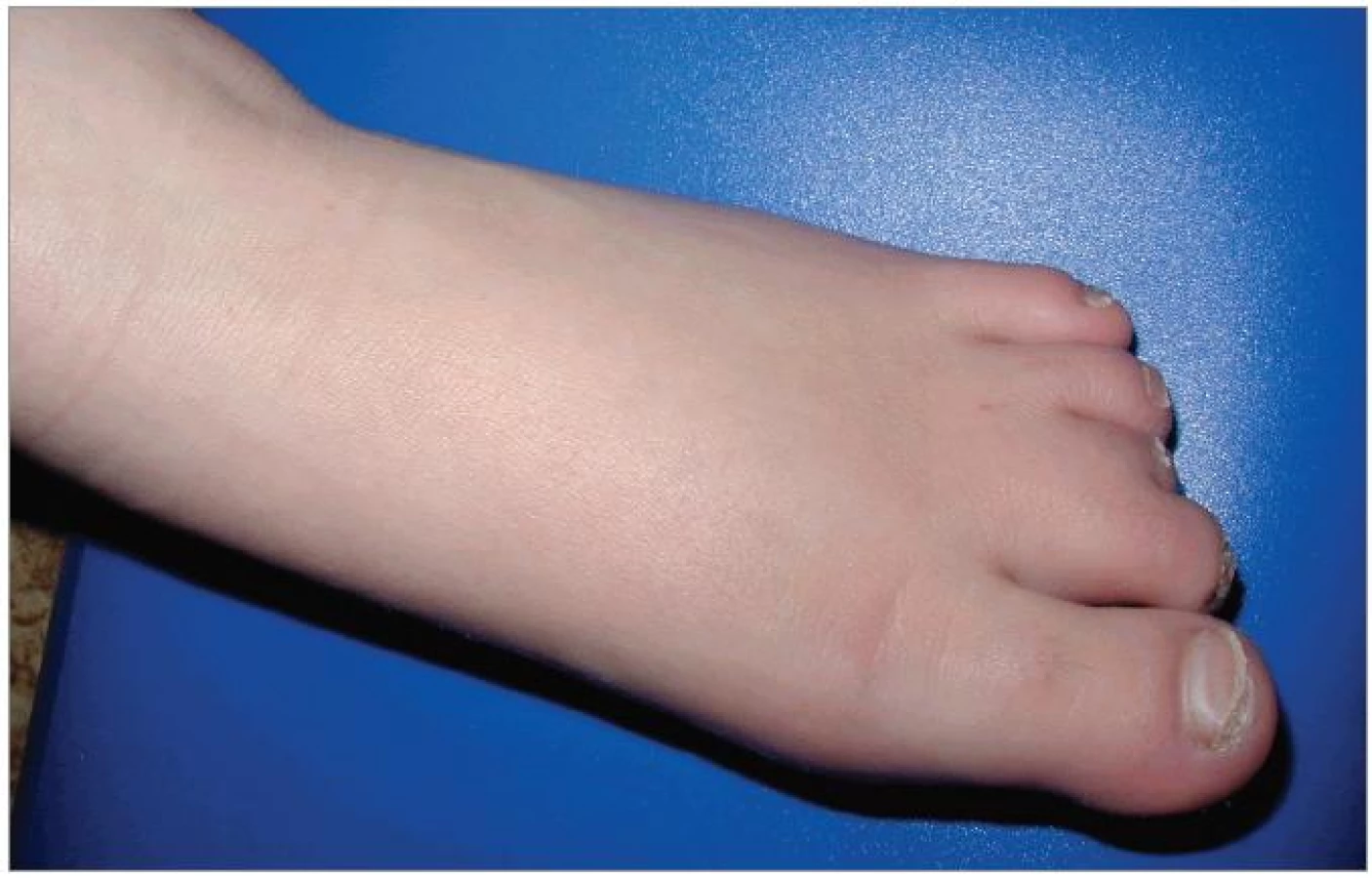
Pomocná vyšetření neprokázala žádné změny krevního obrazu, biochemických parametrů ani hormonů štítné žlázy. EEG záznam byl bez odchylek. CT mozku zobrazilo prostornější komorový systém. Na sonografii břicha bylo patrno jen mírné zvětšení sleziny. Kardiologické vyšetření bylo normální.
Noční polysomnografie zjistila zkrácenou latenci usnutí (3 min), snížené zastoupení REM spánku (16,8 %) a zmnoženou noční bdělost (14 %). Byla patrna opakovaná probouzení a probouzecí reakce z NREM 2 a REM spánku. Delší úsek bdělosti byl zaznamenán mezi 3:45 a 4:30, s opětovným usnutím do 6 h (obr. 3). Celková doba spánku byla 7 hodin. Efektivita spánku byla snížená (84 %). Test mnohočetné latence usnutí prokázal vzhledem k věku mírně zkrácenou průměrnou latenci usnutí (13,4 min). Podle aktigrafického záznamu za 12 dní byla pravidelně patrná kratší doba nočního spánku, usínání nastupovalo mezi 21.–22. hodinou, probouzení mezi 4:00–5:30. Během dne usínala odpoledne nebo k večeru na 1–1,5 hodiny (obr. 4).


Vzhledem k této výrazné poruše cirkadiánní rytmicity jsme pacientku odeslali k opakovanému genetickému vyšetření. Pro podezření na syndrom Smithové-Magenisové bylo indikováno chromozomální vyšetření s vyšší rozlišovací schopností metodou HRT (high resolution technique). Vyšetření prokázalo karyotyp 46, XX, del [17] (p11.2p11.2), a tedy deleci krátkého raménka chromozomu 17, která je pro tento syndrom typická.
Byla zahájena léčba melatoninem (6 mg ve 20 hodin) a acebutololem (400 mg v 8 hodin). Při této léčbě došlo k částečnému zlepšení denní spavosti a kontinuity nočního spánku.
Diskuse
K podezření na diagnózu syndromu Smithové- Magenisové vede charakteristický klinický obraz a kraniofaciální dysmorfie. Stanovení diagnózy nastává většinou v předškolním a školním věku, kdy dochází k plnému rozvoji příznaků, do té doby může být klinický obraz nenápadný. V kojeneckém věku jsou děti hypotonické, pro poruchu orofaciální koordinace mají potíže s příjmem potravy, neprospívají, jsou nápadně klidné, až apatické, což je zčásti podmíněno zvýšenou denní spavostí. Tyto projevy a určitá kraniofaciální dysmorfie vedou většinou, stejně jako u naší pacientky, ke genetickému vyšetření. Vzhled v tomto věku nejčastěji připomíná (epikanty, brachycefalie, široký nos, hypoplazie střední obličejové části) morbus Down, méně často jiné syndromy (Praderové-Williho, velokardiofaciální, Angelmanův syndrom) [5].
Později je patrno opoždění psychomotorického vývoje a výrazné opoždění rozvoje řeči, zejména v expresivní složce. Opožděný rozvoj řeči je v 68 % případů spojen s poruchou sluchu (častěji převodního typu). Mentální retardace je většinou mírného stupně, ale může být i těžká (IQ 20–78, nejčastěji 50–60) [6]. V předškolním a školním věku se dostávají do popředí poruchy chování, poruchy spánku a vyvíjejí se typické kraniofaciální rysy.
Nápadné jsou poruchy chování, které mají ráz sebezraňování, často až automutilačního typu (bouchání hlavou, okusování nebo strhávání nehtů, zasunování předmětů do tělních otvorů). Objevují se záchvaty vzteku a agresivity, které se střídají s velkou emoční příchylností. Děti bývají výrazně hyperaktivní a nesoustředěné. Tendence k stereotypním návykům někdy vede k podezření na autizmus. Dlouho bývají problémy s udržováním čistoty [5,7].
Velmi specifickým projevem, který v tomto případě vedl ke správné diagnóze, je porucha cirkadiánního spánkového rytmu v důsledku inverzní sekrece melatoninu. Spánek nastupuje v časnějších večerních hodinách, je rušený opakovanými probuzeními a celková doba spánku je kratší pro předčasné ranní probuzení. Někdy tak může vzniknout podezření na syndrom předčasné fáze spánku. Polysomnografické a aktigrafické nálezy uváděné v literatuře jsou v souladu s výsledky u naší pacientky. Tato vyšetření prokazují u většiny pacientů zkrácenou celkovou dobu nočního spánku o 1–2 hodiny vzhledem k věku. U poloviny pacientů je redukován nebo fragmentován REM spánek. Přes den se objevuje zvýšená spavost až imperativního rázu [8–10]. Sledování profilu melatoninu prokázalo, že jeho sekrece nastupuje místo ve večerních hodinách v 6 hodin ráno a maxima dosahuje ve 12 hodin, namísto normálního nočního vrcholu. Během noci je naopak sekrece melatoninu minimální. Bylo zjištěno, že výskyt poruch chování souvisí s redukcí doby nočního spánku a poruchou cirkadiánního rytmu. Záchvaty vzteku a agresivity se objevují zejména v dopoledních hodinách, v období vzestupu sekrece melatoninu, a jsou zřejmě mechanizmem, který slouží k překonávání ospalosti, stejně tak jako hyperaktivita. Naopak vrchol sekrece melatoninu je spojen se spánkem v poledních a odpoledních hodinách. Tendence k večernímu časnému usínání odpovídá dalšímu vzestupu sekrece melatoninu mezi 18.–20. hodinou [10,11].
Souvislost genetické poruchy se změněnou sekrecí melatoninu je nejasná. Proti hypotéze o poruše biologických hodin svědčí to, že cirkadiánní profil sekrece kortizolu a růstového hormonu, stejně jako profil teploty, jsou normální. Předpokládá se spíše porucha regulačních systémů, které přímo kontrolují uvolňování melatoninu z pineální žlázy [11].
U pacientky byla rovněž patrná typická kraniofaciální dysmorfie, pro niž je charakteristická brachycefalie, hypoplazie střední obličejové části, široký kořen nosu, epikanty, mohutné a srostlé obočí, prominující dolní čelist, široká ústa. Z postižení dalších systémů byly u pacientky přítomny skeletální abnormity (pes planovalgus) a oční příznaky (šilhání a myopie). Syndaktylie pro tento syndrom není typická, byla podmíněna dědičně (výskyt syndaktylie u matky). Popisovaná je řada dalších poruch a odchylek: jiné oční vady, hypakuze, vrozené vývojové vady srdce a ledvin, rozštěpy rtu a patra, hypogamaglobulinemie, hypercholesterolemie, snížení hladiny tyroxinu, skolióza, růstová retardace. V 75 % případů jsou patrny klinické známky neuropatie – snížená citlivost na bolest a teplo, poruchy chůze, svalová slabost, ale rychlosti vedení periferním nervem jsou normální. Zobrazovací metody (CT, MRI mozku) prokazují nespecifické nálezy, častá je ventrikulomegalie a zvětšená cisterna magna. Až 30 % dětí s tímto syndromem trpí epileptickými záchvaty [6].
Klinická diagnóza byla potvrzena cytogenetickým vyšetřením. Byla detekována intersticiální delece chromozomu 17p11.2. V tomto případě šlo o rozsáhlou deleci viditelnou klasickou analýzou pomocí G-bandingu. Většina pacientů s tímto syndromem má intersticiální deleci menšího rozsahu, která vyžaduje molekulárně cytogenetické vyšetření metodu fluorescenční in situ hybridizace (FISH). Nejčastěji tato chromozomální změna vzniká de novo, avšak k vyloučení familiárního výskytu je indikováno vyšetření obou rodičů. Menší část pacientů má mutaci RAI 1 genu.
Z hlediska léčby se objevují zprávy o příznivém účinku kombinace večerní dávky melatoninu a ranní dávky beta-1 adrenergního blokátoru acebutololu, který potlačuje denní sekreci melatoninu. Někteří autoři při této terapii popisují úpravu spánkového rytmu a podstatné zlepšení chování [12]. U pacientky jsme při podávání doporučených dávek zaznamenali částečné zlepšení kontinuity spánku a výkyvů nálady. Ostatní léčba bývá nespecifická – stabilizátory nálady (valproát, lithium), antipsychotika (risperidon) a inhibitor zpětného vychytávání serotoninu (fluoxetin).
Závěr
Výskyt mentální retardace (většinou lehkého stupně) v kombinaci s poruchami chování autoagresivního rázu a poruchou spánku u dětí s dysmorfickými kraniofaciálními rysy vede k podezření na syndrom Smithové-Magenisové. Velmi specifickým projevem je porucha cirkadiánní rytmicity se zkrácením nočního spánku, předčasným ranním probuzením a zvýšenou denní spavostí. Syndrom je pravděpodobně poddiagnostikován, děti jsou vyšetřovány pro mentální retardaci spojenou s jinými genetickými syndromy či pro autizmus. Potvrzení diagnózy přináší genetické vyšetření. Nadějné se jeví možnosti léčebného ovlivnění abnormní diurnální sekrece melatoninu.
MUDr. Iva Příhodová
Neurologická klinika 1. LF UK a VFN v Praze
Kateřinská 30
128 08 Praha 2
iprih@lf1.cuni.cz
Přijato k recenzi: 6. 11. 2007
Přijato do tisku: 10. 1. 2008
Sources
1. Smith AC, McGavran L, Robinson J, Waldstein G, Macfarlane J, Zonona J et al. Interstitial deletion of (17)(p11.2p11.2) in nine patients. Am J Med Genet 1986; 24: 393–414.
2. Greenberg F, Guzzetta V, Montes de Oca-Luna R, Magenis RE, Smith AC, Richter SF et al. Molecular analysis of the Smith-Magenis syndrome: a possible contiguous-gene syndrome associated with del(17)(p11.2). Am J Hum Genet 1991; 49: 1207–1218.
3. Juyal RC, Figuera LE, Hauge X, Elsea SH, Lupski JR, Greenberg F et al. Molecular analysis of 17p11.2 deletion in 62 Smith-Magenis syndrome patients. Am J Hum Genet 1996; 58: 998–1007.
4. Slager RE, Newton TL, Vlangos CN, Finucane B, Elsea SH. Mutation in RAI 1 associated with Smith-Magenis syndrome. Nat Genet 2003; 33: 466–468.
5. Gropman AL, Duncan WC, Smith AC. Neurologic and developmental features of the Smith-Magenis syndrome (del 17p11.2). Pediatr Neurol 2006; 34: 337–350.
6. Greenberg F, Lewis RA, Potocki L, Glaze D, Parke J, Killian J et al. Multi-disciplinary clinical study of Smith-Magenis syndrome (deletion 17p11.2). Am J Med Genet 1996; 62: 247–254.
7. Smith AC, Dykens E, Greenberg F. Behavioral phenotype of Smith-Magenis syndrome (del 17p11.2). Am J Med Genet 1998; 81: 179–185.
8. Smith ACM, Dykens E, Greenberg F. Sleep disturbance in Smith-Magenis syndrome (del 17p11.2). Am J Med Genet 1998; 81: 186–191.
9. Potocki L, Glaze D, Tan DX, Park SS, Kashork CD, Shaffer LG et al. Circadian rhythm abnormalities of melatonin in Smith-Magenis syndrome. J Med Genet 2000; 37: 428–433.
10. De Leersnyder H, de Blois MC, Claustrat B, Romana S, Albrecht U, Von Kleist-Retzow JC et al. Inversion of the circadian rhythm of melatonin in the Smith-Magenis syndrome. J Pediatr 2001; 139: 111–116.
11. De Leersnyder H, de Blois MC, Vekemans M, Sidi D, Villain E, Kindermans C et al. Beta1-adrenergic antagonists improve sleep and behavioural disturbances in a circadian disorder Smith-Magenis syndrome. J Med Genet 2001; 38: 586–590.
12. De Leersnyder H, Bresson JL, de Blois MC, Souberbielle JC, Mogenet A, Delhotal-Landes B et al. Beta 1-adrenergic antagonists and melatonin reset the clock and restore sleep in a circadian disorder, Smith-Magenis syndrome. J Med Genet 2003; 40: 74–78.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2008 Issue 2
Most read in this issue
- Multiple sclerosis
- Hemangioblastoma and its treatment using Leksell Gamma Knife
- Treatment Results of Low-Grade Gliomas in Children (a Retrospective Data Analysis)
- Smith-Magenis syndrome: a case report