
Roztroušená skleróza
Multiple sclerosis
Multiple sclerosis (MS) is a multifocal inflammatory disease of the CNS in young adults the pathogenesis of which involves autoimmune and neurodegenerative mechanisms. Both factors and a combination of exogenous factors play a role in the etiology of the disease. Inflammatory and therefore partially controllable processes prevail at initial stages. The clinical picture at later stages depends on the degree of axonal loss, and even though it depends on inflammatory tissue destruction to a great extent, it is also partially inflammation-independent. New diagnostic criteria have accelerated the diagnostic process today. MS is not curable, but since 1993 there have been drugs available which influence the natural course of the disease, i.e. beta interferon and the more recent glatiramer acetate. The effect of the above drugs is most potent if treatment starts at the earliest possible stages of the disease. Data have been accumulating about the ability of the drugs to delay the advance of the disease if treatment starts at onset of the initial clinical symptoms (the clinically isolated syndrome – CIS). In case of insufficient therapeutic response to the first choice drugs or if the disease starts with a high inflammatory activity, the use of natalizumab, a recently introduced monoclonal antibody, is indicated. Treatment can be escalated also with the use of cytostatics or even by immunoablation with the support of autologous haematopoetic stem cells. Symptomatic treatment and rehabilitation form integral part of comprehensive care for MS patients which should be provided by a multidisciplinary team. Effective care relieves great part of MS patients of the burden of fatality associated with the disease for decades.
Key words:
multiple sclerosis – diagnostic criteria – beta interferon – glatiramer acetate – natalzumab
:
E. Havrdová
:
Neurologická klinika 1. LF UK a VFN, Praha
:
Cesk Slov Neurol N 2008; 71/104(2): 121-132
:
Minimonography
Roztroušená skleróza (RS) představuje víceložiskové zánětlivé onemocnění CNS mladých dospělých s podílem autoimunitních i neurodegenerativních dějů v patogenezi. V etiologii se uplatňují jak genetické faktory, tak kombinace faktorů v prostředí. V prvních stadiích převažují děje zánětlivé, tedy i léčebně parciálně ovlivnitelné. Klinický stav v pozdějších stadiích je určen mírou axonální ztráty, která je sice z velké části důsledkem destrukce tkáně způsobené zánětem, ale do určité míry je na zánětu i nezávislá. Diagnostiku urychlila nová verze diagnostických kritérií. Onemocnění není vyléčitelné, od r. 1993 jsou však k dispozici léky ovlivňující přirozený průběh nemoci – interferon beta, později glatiramer acetát. Tyto léky mají nejvyšší účinnost, je-li léčba zahájena v co nejčasnějších stadiích nemoci. Přibývá dat o jejich schopnosti oddálit vývoj nemoci, je-li léčba zahájena v době prvních klinických příznaků (klinicky izolovaného syndromu – CIS). Pokud není dostatečná terapeutická odpověď na léky první volby nebo onemocnění začíná s vysokou aktivitou častých atak s nedostatečnou úzdravou, je indikováno podávání natalizumabu, monoklonální protilátky nedávno uvedené do léčebné praxe. Léčbu lze eskalovat i podáním cytostatik, event. imunoablativní léčbou s podáním autologních hematopoetických kmenových buněk. Neoddělitelnou součástí léčby je symptomatická terapie, rehabilitace a režimová opatření. Péči o pacienta by měl zajišťovat multidisciplinární tým. Komplexní péče zbavuje pro velkou část pacientů RS osudovosti, která ji provázela po desetiletí.
Klíčová slova:
roztroušená skleróza – diagnostická kritéria – interferon beta – glatiramer acetát – natalizumab
Úvod
Roztroušená skleróza mozkomíšní (RS) přestala být v posledních 15 letech tématem tabuizovaným a neoblíbeným, a to především pro nové možnosti nejen diagnostiky (s příchodem a širokým využitím MRI), ale především s úsvitem éry nových léčebných možností. Ty byly zpočátku přijímány neurology s velkou nedůvěrou. Zčásti možná proto, že bez výjimky šlo a jde o léky ovlivňující imunitní systém. S nárůstem kontrolovaných dat o jejich vlivu na přirozený průběh nemoci, který byl donedávna hodnocen jako neovlivnitelný, však přibývá ve výzkumu i klinické praxi těch, kteří se problematice RS věnují.
RS je víceložiskové chronické onemocnění centrálního nervového systému (CNS) [1]. V patogenezi se předpokládá zásadní účast autoimunitních dějů (útok imunitního systému je veden proti antigenům především bílé hmoty CNS, dochází však i k poškození šedé hmoty). Čím je autoimunitní útok vyvolán, není přesně známo, jde zřejmě o kombinaci určité genetické vnímavosti se zevními faktory (stres, recidivující a neléčené infekce vedoucí k aktivaci imunitního systému, změny hladin pohlavních hormonů – především v poporodním období, zřejmě i vliv nadbytku 6-omega nenasycených mastných kyselin a nedostatku vitaminu D v potravě, kouření atd).
Zánětlivé děje, proti kterým jsou namířeny prakticky veškeré léčebné postupy u RS, převažují v prvních letech choroby. V pozdějších stadiích nemoci dominují děje neurodegenerativní, v současné době však není známo, do jaké míry neurodegenerativní proces závisí na zánětu. Zatím lze odvodit z patogenetických poznatků základní a zatím jedinou možnou léčebnou strategii – včasné zahájení protizánětlivé léčby.
Historické poznámky
První systematický popis RS pochází od J. M. Charcota z r. 1860. Ačkoli lze nalézt i ve starší literatuře sporadické poznámky o této nemoci, k Charcotově popisu, včetně patologických změn týkajících se jak myelinu, tak úbytku axonů, se lze přihlásit i dnes. Popis klinických příznaků nemoci lze však najít daleko dříve mimo lékařskou literaturu (severské ságy, sv. Lidwina). Příznaky, které se objevovaly a často zcela mizely, vzbuzovaly vždy především obavy a nejistotu. Stejně iracionální byly léčebné postupy až do poloviny minulého století.
S vývojem imunologie jako oboru a zavedením animálních modelů do studia humánních chorob začala vycházet najevo souvislost imunitních dějů s postižením CNS u RS, zprvu velmi okrajově. Poznatky získané studiem experimentální alergické encefalomyelitidy (EAE) nebyly zprvu dávány do souvislosti s RS vůbec. Mozek byl považován za imunoprivilegovaný orgán, v němž imunitní děje téměř neprobíhají. Teprve druhá polovina 20. století přinesla zásadní poznatky o aktivaci lymfocytů proti antigenům CNS na periferii, vstupu imunitních buněk přes hematoencefalickou bariéru (HEB), rozpoznání antigenu v CNS, úloze makrogáfů a mikroglie. Poznatky posledních 2 dekád minulého století pak již měly zásadní důsledky pro vývoj nových biologických léků, které blokují či aktivují jednotlivé molekuly imunitního systému. Stálým problémem však zůstává intenzivně studovaná problematika degenerace nervových vláken u RS. Je nepochybně z velké části důsledkem škod způsobených zánětem, na druhé straně se zdá, že se odehrává i nezávisle na zánětu (např. u primární progrese RS), dominuje v pozdějších stadiích RS a je jen minimálně ovlivnitelná dostupnými léčebnými metodami [2–4].
Epidemiologie
Největší výskyt RS je u indoevropské rasy v mírném pásmu severní polokoule, tzn. i v ČR, kde činí prevalence 80–130 osob na 100 000 obyvatel. V Evropě a Severní Americe stoupá prevalence se zeměpisnou šířkou, a to především u obyvatel skandinávského původu.
Nejméně se RS vyskytuje na rovníku a u černošského obyvatelstva. V Asii je běžnější varianta demyelinizačního onemocnění CNS – neuromyelitis optica (Devicova nemoc) vedoucí k časné ztrátě zraku a hybnosti dolních končetin. Migrační studie naznačují, že v další generaci mají i osoby z oblasti nízkého výskytu vyšší prevalenci nemoci [1]. To dává tušit významný vliv zevních faktorů (vliv zeměpisné šířky je nejspíše kombinací výskytu určitých virů ovlivňujících vývoj imunitního systému v dětství a nedostatku slunečního záření). Genetický podíl je však také jednoznačný, protože některé etnické skupiny jsou ušetřeny (Eskymáci, Laptěvové, maurské obyvatelstvo Malty apod), a v populacích, kde je výskyt RS 1 : 1000 (0,1 %), má příbuzný 1. stupně nemocného s RS 3–4% riziko vyvinout RS, jednovaječné dvojče 34% riziko (oproti adoptovanému dítěti rodiče s RS, které má riziko stejné jako daná populace 0,1 %).
Onemocnění je diagnostikováno nejčastěji ve věku 20–40 let při vývoji prvních klinických příznaků, může však dlouho probíhat inaparentně, a to tehdy, pokud se ložiska zánětu tvoří pouze v takových oblastech CNS, kde nevyvolají nápadnější klinické příznaky. Jsou ovšem známy i případy dětí s RS, stejně tak jako nově diagnostikovaní pacienti nad 55 let. Rizikovými obdobími jsou období puberty, menarche, menopauzy, poporodní období, stres včetně stresu infekčního. Především časté virové infekce jsou spojovány jak s objevením se prvních příznaků, tak s exacerbací choroby, nepochybně pro svou schopnost aktivovat nespecificky imunitní systém.
Ženy jsou postiženy v průměru 2krát častěji než muži, což může být vysvětleno vlivem pohlavních hormonů na imunitní systém.
Průběh nemoci je u 85 % pacientů v prvních 10–15 letech relabující remitující, s různě dlouhými obdobími remisí a s různou mírou trvalých následků po jednotlivých atakách (relapsech). Pak přechází onemocnění do stadia sekundární chronické progrese, kdy zvolna postupuje neurologická invalidita (za nebo bez přítomnosti akutních atak nemoci). Je to stadium vyčerpání axonálních rezerv CNS (většinou od stadia Kurtzkeho EDSS škály 4–4,5, při kterém je dosah chůze bez opory a bez zastavení omezen na 500–300 m). Asi 15 % pacientů má pozvolný nárůst neurologické invalidity od počátku nemoci, především v podobě spastické paraparézy dolních končetin. Tento typ onemocnění, označovaný jako primární progrese, má patogeneticky poněkud odlišný obraz – více neurodegenerace, ztráty oligodendrocytů i axonů než zánětu. Proto je také léčebně zatím málo ovlivnitelné. Asi 3 % pacientů mají maligní (relabující progredující) průběh choroby, charakterizovaný těžkými atakami a progresí neurologického nálezu s časnou těžkou invaliditou.
Termín „benigní RS“ je někdy užíván v případě mnohaletého průběhu choroby bez atak či nárůstu invalidity. V současnosti však nemáme k dispozici žádné spolehlivé metody, jak takový průběh RS předem odhalit, a nemůžeme nikdy vyloučit možnou aktivaci či maligní zvrat vývoje choroby. Označení „benigní RS“ proto může být velmi zavádějící a v žádném případě by nemělo vést k nedostatečnému sledování aktivity choroby a oddálení případné léčby, je-li na místě.
Patofyziologie
Imunitní děje v CNS dlouho unikaly pozornosti. Mozek byl považován za imunoprivilegovaný orgán, tedy za orgán, v němž neprobíhá surveillance (dohled) imunitního systému. Tento dojem byl odvozen z řady dílčích poznatků, jako jsou např. hematoencefalická bariéra, nízký endotel mozkových cév s nevelkou základní expresí adhezivních molekul, které jsou nutnou podmínkou homingu imunitních buněk v orgánech, minimální přítomnost imunitních buněk v mozkomíšním moku v porovnání s periferní krví, nepřítomnost lymfatických uzlin a lymfatické drenáže CNS. V posledních 2 desítkách let však bylo zjištěno, že ačkoli průnik imunitních buněk do CNS je obtížnější než do jiných orgánů, není zdaleka nemožný. Ukázalo se, že aktivovaný lymfocyt, bez ohledu na antigenní specificitu, je schopen svou produkcí prozánětlivých cytokinů aktivovat endotel k vyšší expresi adhezivních molekul, adherovat k němu a produkcí proteolytických enzymů si „rozpustit“ cestičku přes bazální membránu do tkáně CNS. Produkcí zánětlivých cytokinů (TNF-alfa, interferon gama) aktivuje gliové buňky, které jsou schopny za těchto okolností fungovat i jako pomocné buňky imunitního systému (mikroglie je schopna se přeměnit na plnohodnotnou antigen-prezentující buňku, astrocyt je schopen exprese MHC molekul II. třídy). Ne všechny aktivované lymfocyty, které proniknou do CNS, jsou však schopny zahájit v mozku imunitní reakci vedoucí k poškození tkáně. Jsou to pouze lymfocyty, které jsou aktivovány proti antigenům nacházejícím se ve tkáni CNS. Ty ostatní mozek opět opustí bez způsobení škody. Lymfocyty, které svůj antigen v CNS naleznou, jsou jím reaktivovány a spustí kaskádu dějů vedoucích ke vzniku zánětlivého ložiska.
V zánětlivém ložisku RS (place) dochází jednak k lokální poruše hematoencefalické bariéry (tudy pronikají další zánětlivé buňky do CNS, ty už nemusí být aktivovány specifickým antigenem přítomným v CNS), jednak k destrukci myelinu i samotných nervových vláken a retrográdně při jejich zničení i nervových buněk. Samotné nervové buňky mohou být poškozeny i korovými lézemi, jejichž původ není zcela objasněn (uplatňují se zřejmě toxické látky z likvoru, buněčná infiltrace je v těchto lézích totiž minimální, i když v meningách se tvoří shluky B-lymfocytů podobné uzlinám s dlouhodobou produkcí protilátek) [5,6].
Regenerace nervových vláken v CNS není možná, axonu brání v prorůstání k periferii jednak rychle se vytvářející jizva z astrocytů, jednak zřejmě i nedostatek vhodné sekvence růstových faktorů. Ztráta vláken je tak nevratná a vede při určité míře poškození k trvalé invaliditě.
Oprava zničeného myelinu je možná především v počátečních fázích nemoci, pokud není zničeno příliš mnoho oligodendrocytů, jediných buněk v organizmu schopných tvořit centrální myelin. Určitou měrou se na regeneraci podílejí i prekurzory oligodendrocytů, které lze najít i v dospělém mozku a které jsou v přítomnosti vhodných signálů schopny cestovat (až na 10 mm), vyzrát a při dobře načasovaném doteku s obnaženým vláknem schopny začít je obalovat myelinem. Myelinové úseky vytvořené v rámci regenerace jsou kratší a tenčí, obnova vedení vzruchu proto není zcela dokonalá. Po opakované zánětlivé destrukci v téže lokalitě se regenerační schopnosti oligodendrocytů vyčerpávají. V různé míře v lézi hynou. Signály spouštějící regeneraci myelinu jsou také nedostatečné v případě trvalé a značné intenzity zánětu.
Mechanizmy degenerace nervových vláken se v období chronické progrese uplatňují i nezávisle na zánětu, který se omezuje na aktivní okraje plak a difuzní přítomnost zánětlivých buněk ve tkáni. Kromě zvýšeného influxu kalcia do nervových vláken, zvýšené nabídky glutamátu na synapsi a porušené interakce axon-glie jsou ve hře jistě i další, zatím neznámé mechanizmy, kterým se neumíme účinně bránit. Individuální míra a rychlost ničení nervových vláken však nepochybně rozhoduje o aktuálním stavu pacienta [7].
Klinický obraz a průběh
Akutní vznik neurologických příznaků je dán demyelinizací centrálních drah, tedy kondukčním blokem, který způsobí výpadek funkce. O typu klinických příznaků rozhoduje místo CNS, kde se vytvořil zánětlivý infiltrát (tam, kde jde pohromadě více důležitých drah, objeví se polysymptomatická ataka, v oblasti kolem komor se nemusí klinická symptomatologie projevit vůbec nebo velmi nespecificky) [8]. V průběhu nemoci mají jednotlivá ložiska tendenci splývat. Na rozdíl od jednotlivých ložisek koreluje rozsah splývajících ložisek s tíží neurologické invalidity.
- Jedním z nejčastějších počátečních příznaků je zánět očního nervu – optická neuritida. Projevuje se zamlženým viděním, bolestí při pohybu bulbu, výpadky zorného pole (skotomy typicky centrální), poruchou barevného vidění. Příznaky jsou způsobeny demyelinizací očního nervu, který není nervem periferním, ale výběžkem CNS. Pokud zánět s otokem probíhá v místě průchodu zrakového nervu optickým kanálkem, jsou jeho vlákna ničena zároveň též útlakem. Otok se může přenášet na papilu očního nervu, kde je pak patrna její prominence při vyšetření očního pozadí, častěji je však nález na očním pozadí při optické neuritidě zcela normální („nic nevidí ani pacient ani lékař“). Edém po odeznění zánětu může zanechat parciální nablednutí (atrofii) papily především temporálně, a to i bez následné poruchy zraku. Úprava zraku nemusí být úplná, mohou přetrvat skotomy, poruchy barvocitu, vzácně slepota. Jediné reziduum po optické neuritidě může často představovat Uhthoffův fenomén, kdy při větší zátěži organizmu (horko, fyzická zátěž) může dojít k přechodnému zhoršení vizu, který se opět upraví po odeznění vyvolávajícího vlivu. V takovém případě se nejedná o ataku RS, ale o projev nedostatečného vedení dříve poškozeného nervu v situacích náročnějších na rychlost vedení nervových impulzů.
- Poruchy citlivosti (hypestezie, parestezie, hyperestezie bez typické periferní distribuce) jsou dalším častým iniciálním příznakem RS. Jsou často bagatelizovány nebo přičteny poruchám krční či bederní páteře, a to i u mladých jedinců bez poruchy dynamiky páteře. Vzhledem k tomu, jak je důležitá včasná diagnostika, je nutno na tento problém upozornit. Často tyto obtíže během několika týdnů vymizí a pacient ani nenavštíví lékaře. Lokalizace těchto poruch je jednoduchá, pouze pokud se objevují na trupu a mají určitou hranici. Jinak mohou být způsobena ložiskem kdekoli v průběhu senzitivní dráhy.
- Závažnějšími příznaky jsou centrální poruchy hybnosti (parézy), které jsou provázeny spasticitou (zvýšeným svalovým napětím z přerušení tlumivých drah z vyšších etáží a míšních interneuronů), vyššími reflexy a přítomností pyramidových iritačních jevů. Jsou způsobeny ložisky v průběhu především pyramidové dráhy. Během průběhu choroby se parézy různě kombinují (mono-, hemi-, para- i kvadruparéza) a vedou k závažné hybné invaliditě, především v pozdějších stadiích RS.
- Z postižení mozkových nervů se velmi často objevuje internukleární oftalmoplegie a další poruchy okulomotoriky včetně nystagmu. Pacient je vnímá jako oscilopsii, dvojité vidění, které je většinou provázeno pocitem nejistoty v prostoru, méně často rotační závratí, která je typická spíše pro postižení periferního nervu (VIII.). Léze nacházíme v příslušných drahách řídících okulomotoriku, ačkoli např. jednotlivým typům nystagmu nelze většinou přesnou anatomickou strukturu pro klinika přiřadit. I další mozkové nervy mohou být postiženy, a to klinicky periferním typem parézy (nervus facialis, neuralgie trigeminu). Po odstupu z mozkového kmene má periferní nerv ještě několik milimetrů obalu tvořeno centrálním myelinem, proto zde můžeme najít typickou demyelinizační lézi RS. V pozdních stadiích choroby se vyvíjí postižení postranního smíšeného systému, zde jde však většinou o postižení pseudobulbární.
- Vestibulocerebellární poruchy (nejčastěji intenční tremor, dyskoordinace, dysartrie, mozečková skandovaná řeč, poruchy rovnováhy – především nejistota v prostoru a porucha posturálních reflexů, nystagmus) mohou pacienta závažně invalidizovat i při nepřítomnosti těžších paréz.
- Velmi obtěžujícím příznakem RS jsou sfinkterové obtíže (vezikouretrální dyssynergie – urgence, zpožděný start močení, retence moči, inkontinence a kombinace těchto příznaků, obstipace, inkontinence stolice). Častá je jejich souvislost s mírou postižení dolních končetin, ale mohou se vyskytnout samostatně již na začátku choroby. 60 % mužů trpí v průběhu RS erektilní dysfunkcí, u žen jsou sexuální poruchy méně zmapovány. Léze jsou v tomto případě rozesety v průběhu příslušných drah. Málokdy se tyto příznaky objevují izolovaně, mohou však vzácně stát na počátku klinické manifestace choroby (především retence či erektilní dysfunkce).
- Více než polovinu pacientů s RS provází v některé fázi choroby deprese [9]. Bývá to často v době diagnostiky, v období relapsu, ale i zcela nezávisle. Předpokládá se vliv zánětu na serotonergní transmisi a samotná přítomnost zánětlivých cytokinů (především tumor nekrotizujícího faktoru alfa, produktu aktivovaných makrofágů) se zdá být depresogenní. Depresi může podpořit také podání vysoké dávky kortikosteroidů nebo výjimečně i léčba interferonem beta. Depresi a její detekci je třeba věnovat značnou pozornost, protože sebevražednost je u RS několikanásobně zvýšena (především bilanční sebevraždy). Depresi nelze přičíst jednoznačné lokalizaci lézí, různé studie se ve svých závěrech liší (předpokládají především cerebrální lokalizaci ložisek, léze v levé suprainzulární bílé hmotě, regionální asymetrii cerebrálního krevního průtoku v limbickém kortexu).
- 85 % pacientů trpí v průběhu RS patologickou únavou. Ta je způsobena multifaktoriálně a podílí se na ní nejvíce přenos nervového vzruchu menším počtem vláken, z nichž část je chronicky demyelinizována, a dále přítomnost zánětlivých cytokinů a protilátek difuzně v CNS a vliv těchto látek na normální neurotransmisi. Část nervových vláken je zánětem zničena, což je možno pozorovat jako obraz tzv. „černých děr“ (black holes) na T1W obrazech či jako celkovou atrofii CNS.
- Stejné vlivy jako u deprese a únavy se zřejmě uplatňují při vzniku kognitivních poruch, které můžeme najít při použití baterie neuropsychologických testů již na začátku choroby až u 30 % pacientů. V pokročilém stadiu choroby je diagnostikujeme u více než 60 % pacientů. Týkají se především paměti, udržení pozornosti, exekutivních funkcí, méně prostorové orientace. Extrémně vzácná je fatická porucha. Celkový výkon může být zpomalen a je navíc modifikován únavou a případnou depresí. Je však třeba zdůraznit, že samotná atrofie CNS nekoreluje s kognitivním výkonem. Ten je třeba posuzovat baterií specializovaných neuropsychologických testů, mezi atrofii na zobrazovacích metodách a kognitivní deficit nelze nikdy klást rovnítko!
Diagnostika a diferenciální diagnostika (včetně doporučených postupů)
Diagnostická kritéria prošla od 60. let minulého století složitým vývojem souvisejícím se zaváděním pomocných vyšetřovacích metod. První Schumacherova kritéria z 60. let byla kritérii pouze klinickými, pro potřeby začínajících klinických studií bylo třeba implementovat likvorové a elektrofyziologické nálezy (Poser 1983), záhy však byla nedostačující díky rozšíření vyšetření magnetickou rezonancí (MRI). V r. 2001, kdy již bylo jasné, že původní filozofie tajit před pacientem diagnózu co nejdéle je s příchodem léků schopných ovlivnit přirozený průběh choroby neudržitelná, byla vytvořena kritéria McDonaldova [10], umožňující stanovení diagnózy v 1. roce choroby. I ta ale byla znovu upravena v r. 2005 [11] na podkladě rostoucího množství MRI dat (tab. 1). Užití těchto kritérií umožňuje daleko rychlejší stanovení diagnózy RS, není nutno čekat na vývoj další klinické ataky (další zánětlivé ložisko na MRI vzniká podstatně rychleji a dokumentuje diseminaci v prostoru stejně dobře), a je tak možno přistoupit k časnému zahájení terapie, která se v současné době jeví jediným možným způsobem, jak ovlivnit jinak neodvratitelnou invaliditu spojenou s RS.
![Modifikovaná diagnostická kritéria RS z r. 2005 [11].](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image/07801a215614a3bf409ce9a99b630447.png)
Cílem diagnostického procesu je prokázat diseminaci zánětlivého procesu v prostoru CNS a v čase. Opírá se o klinický nález typický pro RS, dále o vizualizaci lézí rozesetých v prostoru CNS pomocí MRI – T2W nebo FLAIR (fluid-attenuated-inversion-recovery s potlačením signálu likvoru) hyperintenzních ložisek (obr. 1–3), o vyšetření mozkomíšního moku (průkaz zánětlivých dějů: výpočet intratékální syntézy IgG, nález alespoň 2 oligoklonálních pruhů v likvoru, které nejsou přítomny v séru, zvýšený počet lymfocytů a monocytů, průkaz plazmatických buněk), ev. v případě pochybností o evokované potenciály (především prodloužení latence vizuálních EP). V případě nejasných nálezů je třeba vyloučit jiné choroby, které by nález mohly vysvětlit.
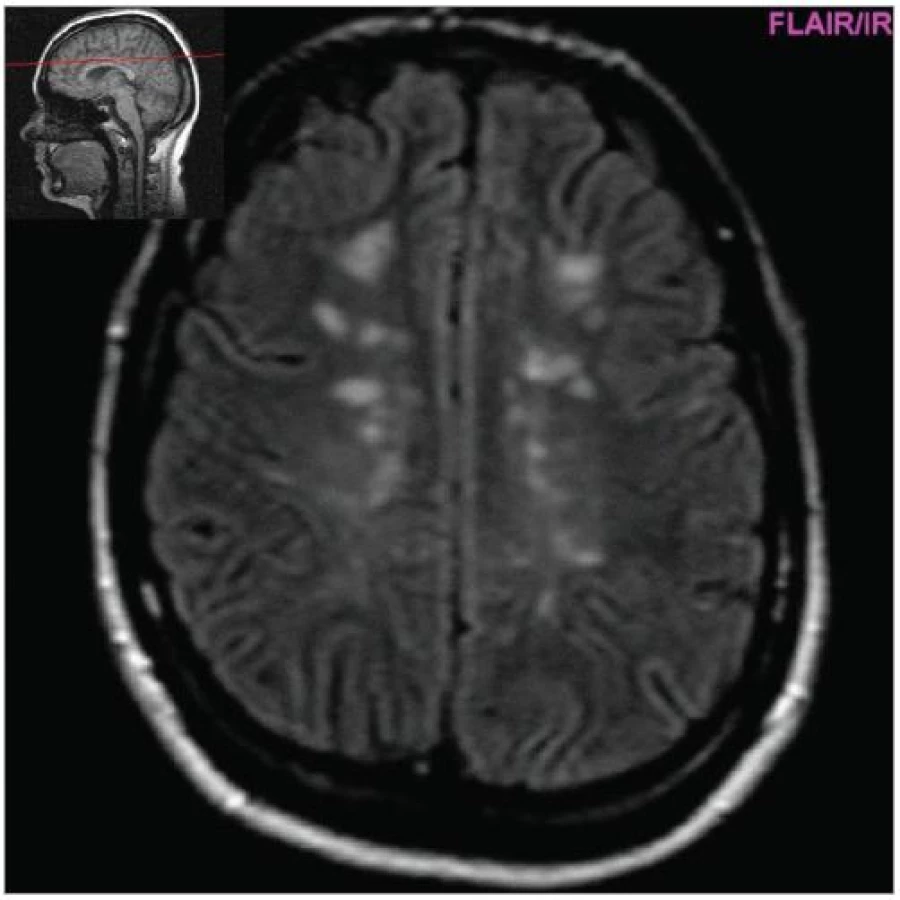
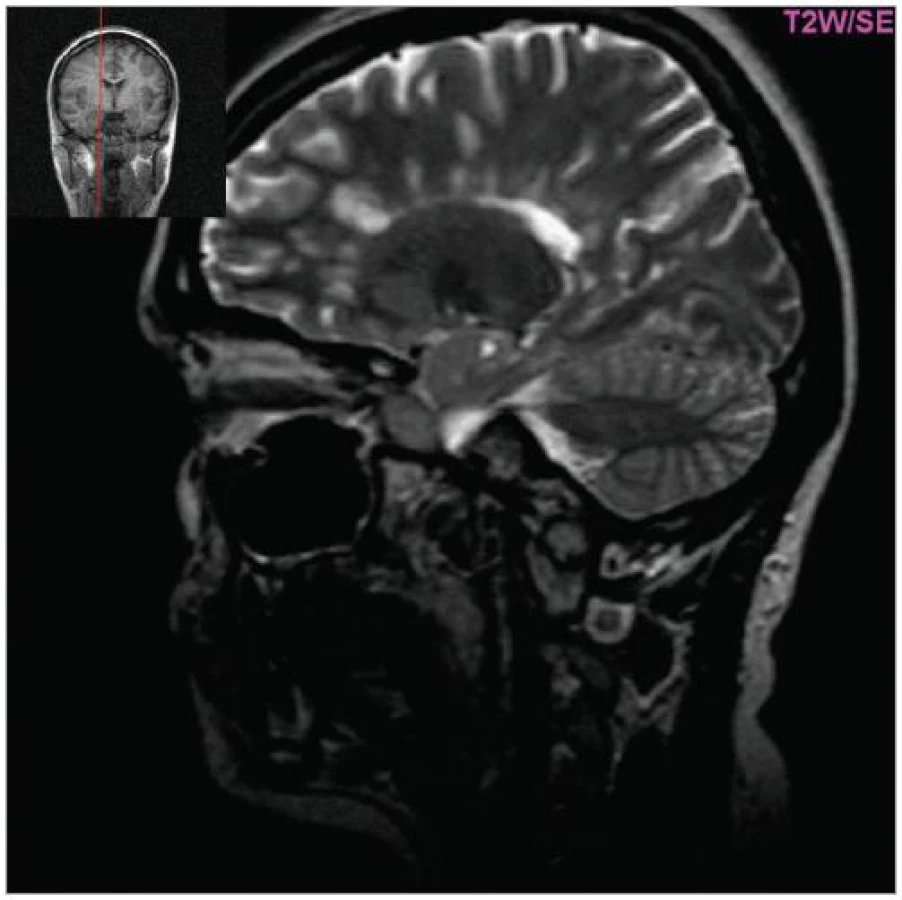
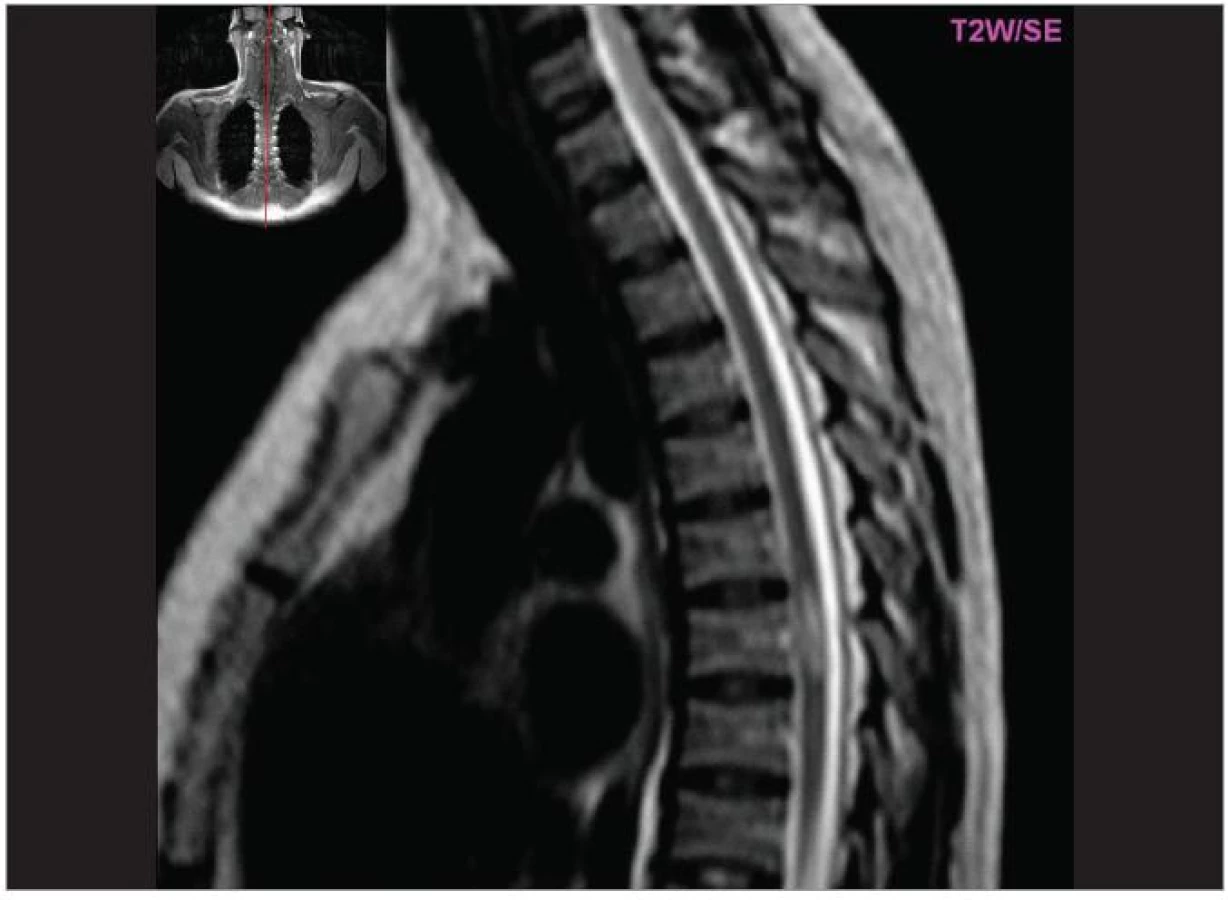
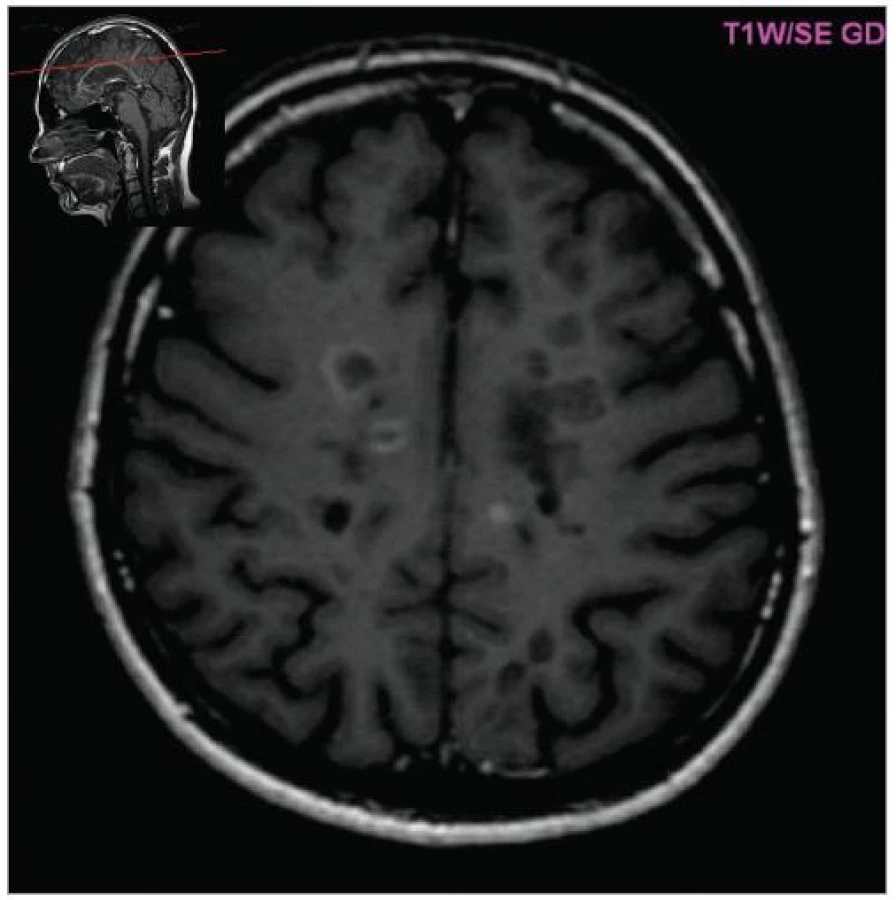
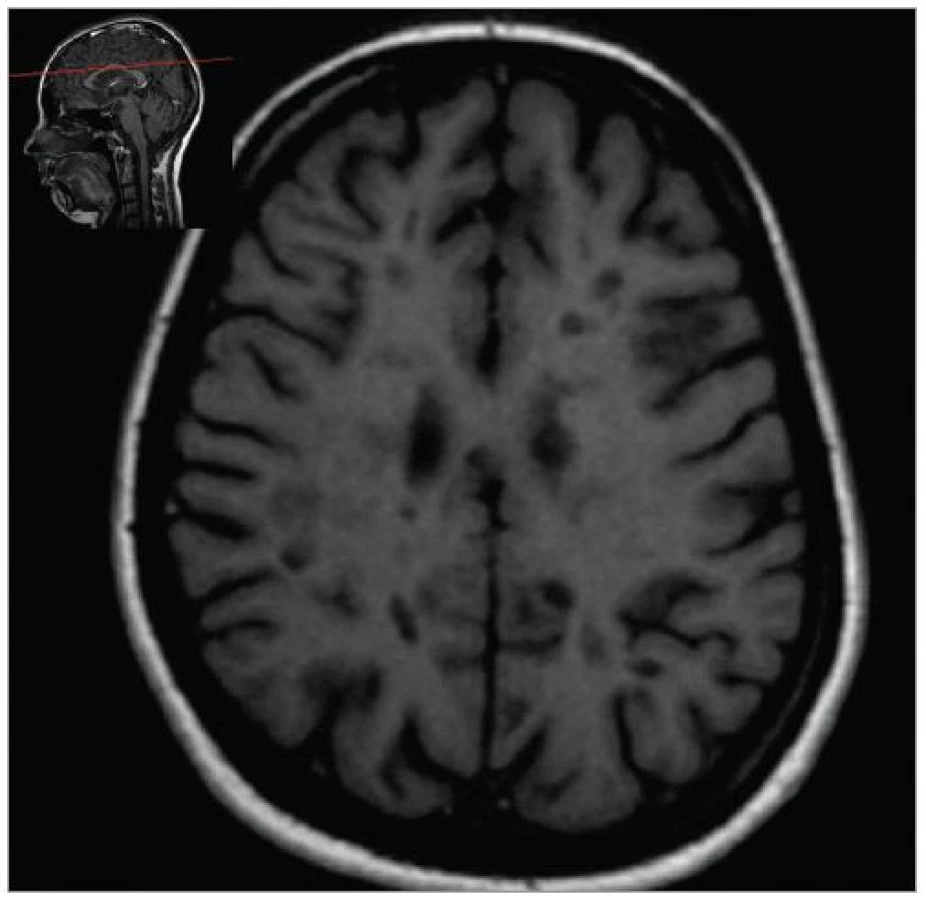
MRI nám navíc umožňuje vizualizovat akutní zánětlivá ložiska v podobě hypointenzních lézí vychytávajících nitrožilně podané gadolinium, které přestupuje v místě zánětu přes porušenou hematoencefalickou bariéru (obr. 4). Můžeme tak hodnotit zánětlivou aktivitu procesu. Nakolik dochází v akutním ložisku k přímé destrukci nervových vláken a nejen k zánětlivé demyelinizaci, nejsme schopni v akutní fázi ataky zjistit, při nedostatečné úzdravě z ataky je však určitá ztráta axonů nepochybná a koreluje s nárůstem atrofie CNS (obr. 5 a 6) a vývojem akutních zánětlivých lézí v perzistující „černé díry“ (hypointenzní ložiska, která již nepředstavují akutní edematózní stadium zánětu, obr. 7).
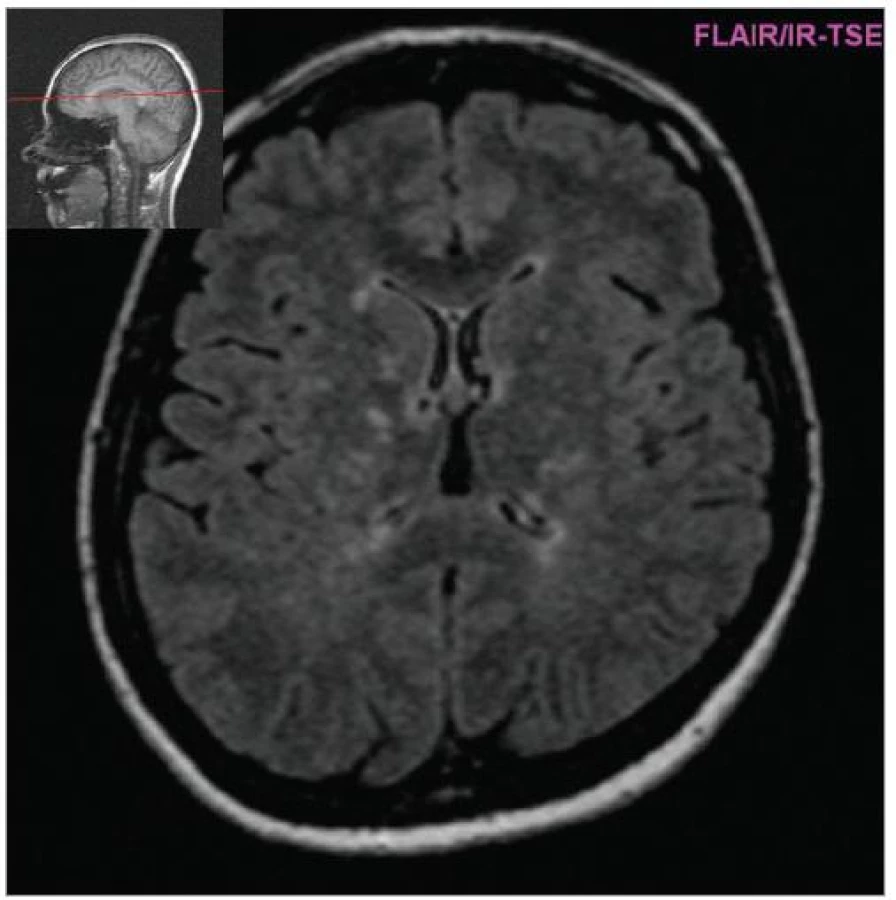
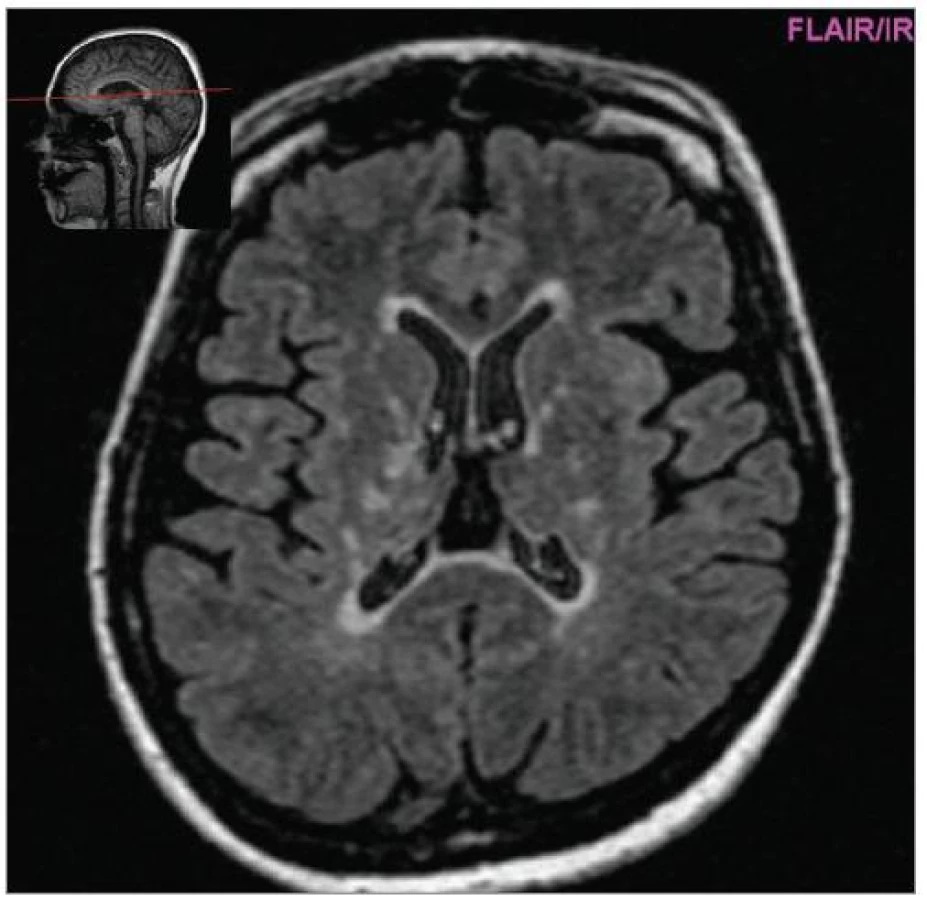
Diferenciální diagnostika [8] je nejbohatší u primárně-progresivního průběhu RS, kde zahrnuje řadu neurodegenerativních chorob a poruch metabolizmu (spinocerebellární ataxie, adultní formu adrenoleukodystrofie, mitochondriální poruchy apod). Většinu ostatních řešitelných možností (nádory mozkového kmene, míšní nádory, rozsáhlejší cévní malformace) odhalí zobrazovací metody. Jiné autoimunity postihující CNS: systémový lupus erytematodes, vaskulitida CNS, Behçetova choroba, mohou být odhaleny pečlivou anamnézou, dalšími příznaky a pomocnými vyšetřeními (autoprotilátky, jiná distribuce hyperintenzních ložisek na MRI, postižení dalších orgánů).
Léčba (včetně dostupných doporučených postupů)
RS není onemocněním vyléčitelným, na druhou stranu v posledních 20 letech značně vzrostla naše možnost chorobu ovlivnit v jejím dlouhodobém průběhu [12,13].
Ovlivnění imunopatologických dějů u RS můžeme rozdělit na:
- léčbu akutní ataky onemocnění
- dlouhodobou léčbu ke snížení počtu atak a zpomalení progrese nemoci
- intenzivní imunomodulační léčbu v případě agresivního průběhu nemoci
- léčbu v případě klinicky izolovaného syndromu suspektního z RS (CIS).
Ad 1. Akutní ataku RS léčíme vysokými dávkami kortikosteroidů, většinou i.v. v celkové dávce kolem 3–5 g. Za zlatý standard se považuje použití metylprednisolonu, u něhož jsou nejvíce potlačeny mineralokortikoidní vedlejší účinky. Pacienta preventivně chráníme před vedlejšími účinky kortikoidní léčby. K těmto opatřením patří ochrana zažívacího traktu (blokátory protonové pumpy, H2 receptorů, antacida) u disponovaných jedinců; prevence osteoporózy (především při častém nebo dlouhodobém podávání u osob s těžkou poruchou hybnosti a u žen v menopauze: D vitamin, magnezium, kalcium, dostatek pohybu, ev. hormonální substituce). U pacientů s diabetem je nutno sledovat glykemii, zpřísnit dietu a dle potřeby navýšit terapii perorálními antidiabetiky či inzulinem.
Je nutné zdůraznit, že čekání, zda příznaky ataky nezmizí bez léčby, je neobhajitelné ve světle medicíny založené na důkazech [14]. Akutní zánětlivé ložisko vzniká několik týdnů, je provázeno poklesem MTR (magnetization transfer ratio) na MRI, pak teprve dochází k vychytávání gadolinia v ložisku. Jestliže je podán pulz metylprednisolonu ihned při nálezu nově enhancující léze, je naděje zachránit část postižené tkáně. I pokud je pacient v této době na dlouhodobé léčbě léky první volby, poškození tkáně je bez akutně podaného metylprednisolonu větší. Efekt kortikoidů není krátkodobý, jak se dlouho tradovalo. Efekt v MTR sekvenci je v dané lézi patrný ještě po 19 měsících. Není proto žádný důvod vyčkávat či léčbu oddalovat. Ve většině případů není pacienta k této léčbě nutno hospitalizovat, ambulantní podání umožňuje daleko větší pružnost v rychlém terapeutickém zásahu.
Ad 2. Dlouhodobá léčba spočívá v trvalé modulaci imunitního systému, musí být tedy v rovnováze riziko a prospěch pacienta (léky musí mít dostatečnou účinnost a minimum vedlejších účinků).
Léky, které prokázaly od r. 1993 ve dvojitě slepých studiích účinnost na snížení počtu relapsů v remitentním (méně i v chronicko-progresivním) stadiu a odpovídají tedy standardu evidence-based medicine (EBM):
- interferon beta (preparáty Betaferon, Rebif, Avonex), který snižuje aktivaci a proliferaci aktivovaných T-lymfocytů, snižuje propustnost hematoencefalické bariéry, snižuje produkci a efekty interferonu gama [15–19]
- glatiramer acetát (Copaxone), který působí jako neencefalitogenní antigenní podnět konkurující antigenům bílé hmoty (myelinovému bazickému proteinu, proteolipid proteinu apod.) a stimulující tvorbu populace tlumivých lymfocytů, které působí v CNS protizánětlivě [20,21]
Léky obsahující interferon beta a glatiramer acetát jsou označovány za léky první volby.
Monoklonální protilátka natalizumab proti adhezivní molekule α4β1 integrinu (Tysabri) je určena pro léčbu aktivní RS či pro situaci neefektivity léčby výše zmíněnými léky. Toto určení neodpovídá původní klinické studii, která ukázala 2násobnou efektivitu oproti interferonu beta či glatiramer acetátu [22], je však kompromisem regulačních orgánů (EMEA), který vychází vstříc klinické potřebě účinného léku a zároveň má poněkud restriktivní efekt. Ten je dán přísnými bezpečnostními opatřeními nastavenými po výskytu 3 případů progresivní multifokální leukoencefalopatie způsobených JC virem v klinických studiích s natalizumabem [23].
K lékům, které prokázaly podobnou účinnost jako interferon beta či glatiramer acetát, ovšem sumací dat z menších studií v metaanalýzách, je ještě nutno přiřadit:
- intravenózní imunoglobuliny (IVIG) s mnohostranným protizánětlivým efektem [24], daným mimo jiné přítomností přirozených protilátek vůči autoantigenům a antiidiotypů vůči autoprotilátkám, funkční blokádou Fc receptorů na makrofázích a mikroglii, neutralizací prozánětlivých cytokinů a jejich receptorů, inhibicí škod způsobených aktivací komplementu atd. IVIG jsou označovány za léky „druhé volby“ doporučované při neefektivitě či intoleranci léků první volby.
Vzhledem k finanční náročnosti těchto léků jsou u nás stanovena přísná kritéria pro jejich použití a ostatní pacienti jsou odkázáni na klasickou imunosupresi používanou u pacientů s jinými autoimunitními chorobami (azatioprin, metotrexát, mykofenolát mofetil ap) [25]. Pro podání těchto látek existuje řada klinických dat, žádná studie však není dostatečně robustní jako u léků modifikujících onemocnění, a to především pro nezájem farmaceutického průmyslu o tyto léta zavedené léky (finanční náročnost rozsáhlých klinických studií je obrovská). V případě užití byť nízkých dávek cytostatik si klinik musí být vědom nutnosti přísnějšího laboratorního sledování k ochraně pacienta před vedlejšími účinky. Kontroly krevního obrazu a jaterních funkcí by měly být prováděny po 3 a 6 týdnech po zahájení podávání, dále pak po 2–3 měsících. Před zahájením léčby azatioprinem by mělo být provedeno genetické vyšetření na polymorfizmy enzymu TPMT (tiopurinmetyltransferáza), aby se předešlo útlumu kostní dřeně [26].
Ad 3. Při agresivním průběhu choroby je nutno použít i agresivnější metody léčby. Standardní dvojitě slepé, placebem kontrolované studie chybí, neboť je nelze organizovat při jejich finanční nákladnosti na každou otázku klinické praxe. Vodítkem jsou zde publikované studie a individuální přístup k informovanému pacientovi.
Jde především o tzv. eskalační léčbu [27], kde nejdříve přistupujeme ke změně preparátu či zvýšení dávky, pulzy vyšších dávek kortikosteroidů a cytostatik (metylprednisolon v kombinaci s cyklofosfamidem či mitoxantronem) [28–32], při maligním průběhu lze zvážit imunoablativní léčbu s podporou autologních hematopoetických kmenových buněk (CD34+), která je ovšem stále ještě léčbou experimentální s malým rizikem mortality (2–5 % dle pracoviště) [33,34].
Ad 4. Klinicky izolovaný syndrom (clinically isolated syndrom, CIS) suspektní z vývoje v RS představuje v současné době oblast zájmu číslo jedna. Je to z důvodu předpokladu, že co nejčasnější ovlivnění nemoci může vést k nejúspěšnějšímu oddálení trvalé invalidity. Několik studií přesvědčivě doložilo, že léčba pacientů, u kterých se vyvinuly první symptomy RS podpořené pomocnými vyšetřeními, skutečně oddaluje nástup další ataky a rozvoj změn na MRI. Posunutí léčby až po druhé klinické atace (tedy dřívější doložení tzv. klinicky jisté RS) znevýhodnilo takové pacienty ve všech dosud provedených klinických studiích [35–38]. Dřívější obavu, že bude až 50 % pacientů léčeno zcela zbytečně, protože nikdy se u nich RS nevyvine, jednoznačně vyvrátila studie BENEFIT, která ukázala, že již do 2 let od první ataky 85 % pacientů splní kritéria pro RS dle McDonalda.
Jací pacienti jsou pro léčbu léky modifikujícími průběh onemocnění po první atace vhodní i v ekonomicky chudším prostředí? Podle dosud dostupných studií zdvojnásobuje přítomnost oligoklonálních pásů v likvoru u pacientů s nálezem alespoň 2 hyperintenzních ložisek na MRI pravděpodobnost další klinické ataky. Pro vývoj invalidity je nejzávažnějším prognostickým faktorem neúplná úzdrava po první atace, tedy neurologický deficit. Těmto pacientům by se přednostně mělo dostat té nejčasnější léčby, a proto je to zapracováno i v nových kritériích pro léčbu RS pro ČR (která však dosud nemají jednoznačně potvrzenou úhradu zdravotními pojišťovnami).
V jakémkoli stadiu choroby je nutno dbát na úlevu od příznaků pomocí symptomatické léčby [8]. Jde především o ovlivnění spasticity (zvýšeného svalového napětí) centrálními myorelaxancii (baklofen, tizanidin, tetrazepam, tiokolchikosid, lokální aplikace botulotoxinu, intratékální baklofenová pumpa) [39]; deprese (SSRI – blokátory zpětného vychytávání serotoninu); sfinkterových obtíží (anticholinergika, myorelaxancia včetně lokálně aplikovaného botulotoxinu, čistá intermitentní katetrizace apod); neurogenní bolesti (antiepileptika karbamazepin, gabapentin, pregabalin). Mozečkový třes nelze u RS farmakologicky ovlivnit (lze zkusit klonazepam, ale účinné dávky většinou neúnosně zvyšují únavu).
Vždy je třeba základní léčbu doplnit o včasnou léčbu všech infekcí a infekčních fokusů antibiotiky.
K terapii patří také péče o fyzickou kondici a rehabilitace [40], jejíž metody jsou založeny na neurofyziologických poznatcích a využívají plasticitu CNS. Podobně kognitivní trénink se začíná uplatňovat v ovlivnění počínajících kognitivních poruch. Zde se v budoucnu jistě uplatní i kognitiva, zatím ověřená především v počáteční fázi Alzheimerovy nemoci [41]. Stejně tak lze únavu ovlivnit modafinilem, užívaným v léčbě narkolepsie.
V terapeutickém arzenálu RS stále ještě chybí léky neuroprotektivní, které by umožnily chránit CNS od samého počátku nemoci.
K péči o pacienta s RS nedílně patří i psychoterapie, sociální poradenství a ergoterapie [42].
Závěr
Zavedení moderních léků do léčby RS zásadně změnilo prognózu řady pacientů, pro které dříve znamenalo sdělení diagnózy sdělení nepříznivého osudu. Terapie se logicky posouvá do časných stadií nemoci. V klinickém testování fáze 3 je řada nových léků s předpokládaným vyšším účinkem na aktivitu nemoci, než jakou známe z již více než 10letého použití léků první volby modifikujících onemocnění i v ČR (fumarát, fingolimod, laqiunimod, kladribin, alemtuzumab, rituximab apod). U některých z těchto léků je i předpoklad neuroprotektivního účinku.
Přestože je léčba RS nákladná, nelze zapomínat, že jde o chronické, nepreventabilní onemocnění, které je diagnostikováno u většiny nemocných v časné dospělosti, a trvalá invalidita těchto pacientů představuje tedy pro společnost zátěž daleko větší než včasná léčba [43].
MRI dokumentace zapůjčena s laskavým svolením prof. MUDr. Z. Seidla, CSc., Radiodiagnostická klinika 1. LF UK v Praze a VFN Praha.
doc. MUDr. Eva Havrdová, CSc.
Neurologická klinika 1. LF UK a VFN, Praha
Kateřinská 30
120 00 Praha 2
e-mail: ehavr@lf1.cuni.cz
Přijato k recenzi: 30. 1. 2008
Přijato do tisku: 26. 3. 2008
Recenzenti:
doc. MUDr. Pavel Štourač, CSc.
MUDr. Jiří Piťha, CSc.
MUDr. Jan Mareš, Ph.D.
doc. MUDr. Eva Havrdová, CSc.

Eva Havrdová vystudovala Fakultu všeobecného lékařství UK v Praze v r. 1981. Do r. 1996 pracovala ve Fakultní Thomayerově nemocnici v Praze Krči, od r. 1996 na 1. LF UK v Praze na neurologické klinice VFN. V r. 1995 obhájila kandidátskou práci na téma cytokinů a lymfocytárních subpopulací v periferní krvi a likvoru pacientů s roztroušenou sklerózou (RS) a v r. 2003 habilitovala v oboru neurologie. Pod vedením prof. MUDr. P. Jedličky, DrSC. se od začátku své praxe věnovala problematice roztroušené sklerózy, stála u zavádění moderních léků ovlivňujících přirozených průběh RS do klinické praxe v ČR, ve VFN vybudovala největší Centrum pro demyelinizační onemocnění v ČR. Věnuje se výuce studentů 1. LF UK v Praze i postgraduálních studentů, je řešitelkou řady vědeckých národních i mezinárodních grantů. Účastnila se mnoha klinických studií u RS, většinou jako národní koordinátorka. Je členkou řady mezinárodních organizací, v současné době pracuje ve výkonném výboru European Multiple Sclerosis Platform, je předsedkyní Neuroimunologické sekce ČNS ČLS JEP. V roce 2007 předsedala mezinárodnímu kongresu ECTRIMS (European Committee for
Treatment and Research of Multiple Sclerosis) v Praze. Je autorkou mnoha odborných i popularizačních publikací, její kniha Neuroimunologie byla oceněna cenou Hlávkovy nadace a ČLF za nejlepší publikaci v oboru medicíny za rok 2001 a cenou Neurologické společnosti ČLS JEP za nejlepší monografii v oboru neurologie za rok 2001. Maximální úsilí v současné době věnuje jednak rozšíření možností léčby RS od jejího počátku před vznikem neurologické invalidity, jednak experimentální léčbě maligních forem nemoci imunoablací s podporou hematopoetických kmenových buněk.
Vědomostní test
1. Roztroušená skleróza:
- a) prevalence v ČR je 80–130 osob na 100 000 obyvatel
- b) prevalence v ČR je 50–70 osob na 100 000 obyvatel
- c) prevalence klesá se zeměpisnou šířkou směrem na sever
- d) v Asii je RS méně častá než neuromyelitis optica (Devicova nemoc)
2. Roztroušená skleróza:
- a) je autozomálně dominantní onemocnění s mutací na 4. chromozomu
- b) je multifaktoriální onemocnění s podílem genetických a zevních faktorů
- c) v ČR je riziko rozvoje onemocnění u příbuzného prvního stupně pacienta s RS 25%
- d) v ČR je riziko rozvoje onemocnění u příbuzného prvního stupně pacienta s RS 3–4%
3. Mezi riziková období pro rozvoj nebo exacerbaci RS patří:
- a) 1. trimestr gravidity
- b) 2. trimestr gravidity
- c) poporodní období
- d) febrilní infekt
4. Onemocnění RS:
- a) začíná nejčastěji mezi 20.–40. rokem věku
- b) se může manifestovat v dětském věku
- c) v 85 % začíná jako relabující remitující forma
- d) po vyčerpání rezerv CNS přechází do léčebně málo ovlivnitelné sekundární chronické progrese
5. V patogenezi RS:
- a) hrají zcela zásadní roli autoprotilátky proti antigenům CNS
- b) dochází k zánětlivému postižení pouze bílé hmoty CNS (demyelinizaci)
- c) se uplatňuje především buněčná imunita
- d) dochází k postižení bílé i šedé hmoty mozkové
6. Klinické projevy RS:
- a) zraková dráha bývá nejčastěji postižena v oblasti optického nervu (optická neuritis)
- b) konstantní parestezie nepředstavují ataku RS
- c) RS jako onemocnění CNS nemůže způsobit periferní parézu n. facialis
- d) typickou poruchu nálady představuje nepřiměřená euforie
7. V diagnostice RS hraje zásadní roli:
- a) MRI mozku, resp. míchy, analýza mozkomíšního moku, ev. vizuální evokované potenciály
- b) CT mozku, analýza mozkomíšního moku, všechny modality evokovaných potenciálů
- c) MRI mozku, imunologické vyšetření periferních mononukleárních buněk, somatosenzorické evokované potenciály
- d) MRI mozku, resp. míchy, evokované potenciály všech modalit, EMG
8. Magnetická rezonance u RS umožňuje:
- a) ozřejmit ložiska zánětu v CNS (mozku i míchy)
- b) zhodnotit atrofii CNS, která vždy koreluje s kognitivním deficitem
- c) prokázat aktivitu ložiska zánětu s porušenou hematoencefalickou bariérou pomocí podání gadolinia
- d) včasnější stanovení diagnózy RS oproti klinické manifestaci (ložiska na MRI přibývají až 3krát častěji než klinické ataky)
9. Zlatým standardem v léčbě akutní ataky RS je:
- a) p.o. prednisolon (postupné snižování dávky od 40 mg/den)
- b) dexametazon
- c) metylprednisolon do celkové dávky 3–5 g
- d) ACTH
10. Léčbu léky první volby (DMD) optimálně zahajujeme v tomto stadiu onemocnění:
- a) CIS (klinicky izolovaný syndrom)
- b) sekundárně progresivní forma RS, kdy dochází ke klinicky zjevnému nárůstu invalidity
- c) po minimálně 2 atakách motorické symptomatologie za posledních 24 měsíců
- d) u žen s diagnózou RS v poporodním období
11. Mezi léky první volby (DMD) u RS patří:
- a) glatiramer acetát
- b) interferon beta
- c) azatioprin
- d) imunoablativní léčba s podporou autologních hematopoetických kmenových buněk
12. Natalizumab (Tysabri) je:
- a) monoklonální protilátka proti adhezivní molekule a4b1-integrinu
- b) určen pro pacienty s primárně progresivní formou RS
- c) určen pro pacienty s relabující-remitující formou RS, v případě nedostatečné účinnosti dosavadních léků první volby
- d) pro vysoce aktivní relabující-remitující RS
13. Mezi možné nežádoucí účinky léčby kortikosteroidy nepatří:
- a) podráždění žaludeční sliznice až vředová choroba gastroduodenální
- b) osteoporóza
- c) infertilita
- d) hypokalemie
14. Prevence nežádoucích účinků kortikosteroidů zahrnuje:
- a) substituci draslíku
- b) ochranu žaludeční sliznice (H2 antihistaminika, blokátory iontové pumpy)
- c) frekventní kontroly glykemie a případnou úpravu dávky inzulinu u diabetika
- d) substituci vitaminu D a kalcia při dlouhodobém podávání kortikosteroidů
15. Azatioprin:
- a) představuje klasické imunosupresivum, které může způsobit elevaci jaterních enzymů; při jeho léčbě jsou proto nutné pravidelné laboratorní kontroly
- b) v léčbě RS představuje lék první volby
- c) aktivitu enzymu TPMT (tiopurinmetyltransferáza) vyšetřujeme, pokud při terapii azatioprinem dojde k poklesu leukocytů pod 1,0 × 109/l
- d) aktivitu enzymu TPMT vyšetřujeme standardně před zahájením léčby azatioprinem k vyloučení rizika útlumu kostní dřeně spojeného s nízkou aktivitou enzymu
16. Klinicky izolovaný syndrom (CIS):
- a) představuje první klinickou atakususpektní z rozvoje definitivní RS dle pomocných vyšetření (MRI mozku a mozkomíšní mok)
- b) představuje první klinickou ataku suspektní z rozvoje definitivní RS dle pomocných vyšetření, kritéria pro definitivní RS však nejsou splněna a imunomodulační léčba není indikována
- c) je optimální dobou pro zahájení imunomodulační léčby léky první volby
- d) do 2 let od CIS jsou splněna McDonaldova diagnostická kritéria pro RS u 85 % pacientů
17. McDonaldova kritéria:
- a) jsou diagnostická kritéria pro RS založená na klinickém a likvorologickém nálezu
- b) jsou diagnostická kritéria pro RS založená především na magnetické rezonanci
- c) umožňují stanovit diagnózu RS dříve než podle klinické aktivity onemocnění, a tím zahájit včas adekvátní léčbu
- d) podstatou je průkaz diseminace zánětlivého procesu v čase i v prostoru CNS
18. Symptomatická léčba RS:
- a) centrální myorelaxancia indikujeme ve velmi individuální dávce dle subjektivních obtíží pacienta (tuhosti, křeče), nikoliv paušálně pro nález spasticity při klinickém vyšetření
- b) v případě asymptomatické signifikantní bakteriurie u pacienta s RS není na místě přeléčení antibiotiky dle citlivosti
- c) léky volby na depresi u RS představují tricyklická antidepresiva
- d) sfinkterové obtíže se mohou vyskytnout již na začátku choroby a jejich léčba by měla probíhat ve spolupráci se zkušeným neurourologem
19. 24 hodin trvající, konstantní levostranné hemiparestezie u pacienta s RS:
- a) nepředstavují ataku RS
- b) představují ataku RS a je indikována léčba metylprednisolonem
- c) představují ataku RS, vzhledem k přítomnosti pouze senzitivních příznaků není indikována léčba
metylprednisolonem - d) subjektivní senzitivní obtíže léčíme jako ataku až po verifikaci léze somatosenzorické dráhy pomocí evokovaných potenciálů
20. Gravidita u pacientek s RS:
- a) zásadně není doporučována
- b) je nutno optimálně plánovat na dobu stabilizace RS (klinicky i dle magnetické rezonance)
- c) je nutno dodržet bezpečnostní interval mezi vysazením imunosuprese a početím (klasická p.o. imunosupresiva – 3 měsíce, cytostatika – 6 měsíců)
- d) pro prozánětlivý efekt prolaktinu doporučujeme zkrátit kojení na 2–3 měsíce
Sources
1. Havrdová E et al. Neuroimunologie. Praha: Maxdorf 2001: 180–263.
2. Lassmann H, Brück W, Lucchinetti CF. The immunopathology of multiple sclerosis: an overview. Brain Pathol 2007; 17(20): 210–218.
3. Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mörk S, Bö L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med 1998; 338(5): 278–285.
4. Dutta R, Trapp BD. Pathogenesis of axonal and neuronal damage in multiple sclerosis. Neurology 2007; 68(22 Suppl 3): S22–31.
5. Kutzelnigg A, Lassmann H. Cortical lesions and brain atrophy in MS. J Neurol Sci 2005; 233(1–2): 55–59.
6. Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol 2004; 14(2): 164–174.
7. Piťha J, Havrdová E: Axonální patologie u roztroušené sklerózy. Cesk Slov Neurol N 2005; 68/101(3): 154–158.
8. Havrdová E. Roztroušená skleróza. Praha: Maxdorf 2006.
9. Sládková V, Muchová B, Ticháčková A, Mareš J, Urbánek K, Kaňovský P. Výskyt depresí u nemocných s roztroušenou sklerózou mozkomíšní. Cesk Slov Neurol N 2006; 69/102(4): 280–285.
10. McDonald WI, Compston A, Edan G, Goodkin D, Hartung HP, Lublin FD et al. Recommended diagnostic criteria for multiple sclerosis: guidelines from the International Panel on the diagnosis of multiple sclerosis. Ann Neurol 2001;50(1): 121–127.
11. Polman CH, Reingold SC, Edan G, Filippi M, Hartung HP, Kappos L et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the "McDonald Criteria". Ann Neurol 2005; 58(6): 840–846.
12. Paty DW, Hartung HP, Ebers GC et al. Management of relapsing-remitting multiple sclerosis: diagnosis and treatment guidelines. Eur J Neurol 1999; 6(Suppl 1): S1–S35.
13. Karussis D, Biermann LD, Bohlega S, Boiko A, Chofflon M, Fazekas F et al. A recommended treatment algorithm in relapsing multiple sclerosis: report of an international consensus meeting. Eur J Neurol 2006; 13(1): 61–71.
14. Richert ND, Ostuni JL, Bash CN, Leist TP, McFarland HF, Frank JA. Interferon beta-1b and intravenous methylprednisolone promote lesion recovery in multiple sclerosis. Mult Scler 2001; 7(1): 49–58.
15. The IFNB Multiple Sclerosis Study Group. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. I. Clinical results of a multicenter, randomized, double-blind, placebo-controlled trial. Neurology 1993; 43(4): 655–661.
16. The IFNB Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group. Interferon beta-1b in the treatment of multiple sclerosis: final outcome of the randomized controlled trial. Neurology 1995; 45(7): 1277–1285.
17. Jacobs LD, Cookfair DL, Rudick RA, Herndon RM, Richert JR, Salazar AM et al. Intramuscular interferon beta-1a for disease progression in relapsing multiple sclerosis. The Multiple Sclerosis Collaborative Research Group (MSCRG). Ann Neurol 1996; 39(3): 285–294.
18. Simon JH, Jacobs LD, Campion M, Wende K, Simonian N, Cookfair DL et al. Magnetic resonance studies of intramuscular interferon beta-1a for relapsing multiple sclerosis. The Multiple Sclerosis Collaborative Research Group. Ann Neurol 1998; 43(1): 79–87.
19. PRISMS (Prevention of Relapses and Disability by Interferon beta-1a Subcutaneously in Multiple Sclerosis) Study Group. Randomised double-blind placebo-controlled study of interferon beta-1a in relapsing/remitting multiple sclerosis. Lancet 1998; 352(9139): 1498–1504.
20. Johnson KP, Brooks BR, Cohen JA, Ford CC, Goldstein J, Lisak RP et al. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind placebo-controlled trial. The Copolymer 1 Multiple Sclerosis Study Group. Neurology 1995; 45(7): 1268–1276.
21. Filippi M, Rovaris M, Rocca MA, Sormani MP, Wolinsky JS, Comi G. Glatiramer acetate reduces the proportion of new MS lesions evolving into "black holes". Neurology 2001; 57(4): 731–733.
22. Polman CH, O'Connor PW, Havrdova E, Hutchinson M, Kappos L, Miller DH et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med 2006; 354(9): 899–910.
23. Kappos L, Bates D, Hartung HP, Havrdova E, Miller D, Polman CH et al. Natalizumab treatment for multiple sclerosis: recommendations for patient selection and monitoring. Lancet Neurol 2007; 6(5): 431–441.
24. Sorensen PS, Fazekas F, Lee M. Intravenous immunoglobulin G for the treatment of relapsing-remitting multiple sclerosis: a meta-analysis. Eur J Neurol 2002; 9(6): 557–563.
25. Fernández O, Fernández V, De Ramón E. Azathioprine and methotrexate in multiple sclerosis. J Neurol Sci 2004; 223(1): 29–34.
26. Frohman EM, Havrdova E, Levinson B, Slanar O. Azathioprine myelosuppression in multiple sclerosis: characterizing thiopurine methyltransferase polymorphisms. Mult Scler 2006; 12(1): 108–111.
27. Rieckmann P, Toyka KV, Bassetti C, Beer K, Beer S, Buettner U et al. Escalating immunotherapy of multiple sclerosis – new aspects and practical application. J Neurol 2004; 251(11): 1329–1339.
28. Hartung HP, Gonsette R, König N, Kwiecinski H, Guseo A, Morrissey SP et al. Mitoxantrone in progressive multiple sclerosis: a placebo-controlled, double-blind, randomised, multicentre trial. Lancet 2002; 360(9350): 2018–25.
29. Edan G, Miller D, Clanet D, Confavreux C, Lyon-Caen O, Lubetzki C et al. Therapeutic effect of mitoxantrone combined with methylprednisolone in multiple sclerosis: a randomised multicentre study of active disease using MRI and clinical criteria. J Neurol Neurosurg Psychiatry 1997; 62(2): 112–118.
30. Tichá V, Havrdová E, Nováková I, Horáková D. Mitoxantron v léčbě aktivní RS. Cesk Slov Neurol N 2003; 66/99(1): 31–37.
31. Weiner HL, Cohen JA. Treatment of multiple sclerosis with cyclophosphamide: critical review of clinical and immunologic effects. Mult Scler 2002; 8(2): 142–154.
32. Smith DR, Weinstock-Guttman B, Cohen JA, Wei X, Gutmann C, Bakshi R et al. A randomized blinded trial of combination therapy with cyclophosphamide in patients-with active multiple sclerosis on interferon beta. Mult Scler 2005; 11(5): 573–582.
33. Saccardi R, Kozak T, Bocelli-Tyndall C, Fassas A, Kazis A, Havrdova E et al. Autologous stem cell transplantation for progressive multiple sclerosis: update of the European Group for Blood and Marrow Transplantation autoimmune diseases working party database. Mult Scler 2006; 12(6): 814–823.
34. Mezey E, Key S, Vogelsang G, Szalayova I, Lange GD, Crain B. Transplanted bone marrow generates new neurons in human brains. Proc Natl Acad Sci USA 2003; 100(3): 1364–1369.
35. Kinkel RP, Kollman C, O'Connor P, Murray TJ, Simon J, Arnold D et al. IM interferon beta-1a delays definite multiple sclerosis 5 years after a first demyelinating event. Neurology 2006; 66(5): 678–684.
36. Comi G, Filippi M, Barkhof F, Durelli L, Edan G, Fernández O et al. Effect of early interferon treatment on conversion to definite multiple sclerosis: a randomised study. Lancet 2001; 357(9268): 1576–1582.
37. Kappos L, Freedman MS, Polman CH, Edan G, Hartung HP, Miller DH et al. Effect of early versus delayed interferon beta-1b treatment on disability after a first clinical event suggestive of multiple sclerosis: a 3-year follow-up analysis of the BENEFIT study. Lancet 2007; 370(9585): 389–97.
38. Achiron A, Kishner I, Sarova-Pinhas I, Raz H, Faibel M, Stern Y et al. Intravenous immunoglobulin treatment following the first demyelinating event suggestive of multiple sclerosis: a randomized, double-blind, placebo-controlled trial. Arch Neurol 2004; 61(10): 1515–1520.
39. Štětkářová I, Šroubek J, Vrba I, Peregrin J, Havrdová E. Jednorázové intratékální podání baklofenu a následné zavedení pumpového systému v léčbě těžké spasticity u osob s roztroušenou sklerózou. Cesk Slov Neurol N 2007; 70/103(2): 190–195.
40. Kesselring J, Beer S. Symptomatic therapy and neurorehabilitation in multiple sclerosis. Lancet Neurol 2005; 4(10): 643–652.
41. Christodoulou C, Melville P, Scherl WF, Macallister WS, Elkins LE, Krupp LB. Effects of donepezil on memory and cognition in multiple sclerosis. J Neurol Sci 2006; 245(1–2): 127–36.
42. Dušánková J, Havrdová E. Psychiatrická problematika u sclerosis multiplex. Neurol pro praxi 2006; 4: 192–194.
43. Kobelt G, Berg J, Lindgren P, Fredrikson S, Jönsson B. Costs and quality of life of patients with multiple sclerosis in Europe. J Neurol Neurosurg Psychiatry 2006; 77(8): 918–926.
44. Barkhof F, Filippi M, Miller DH et al. Comparison of MRI criteria at first presentation to predict conversion to clinically definite multiple sclerosis. Brain 1997; 120: 2059–2069.
45. Tintoré M, Rovira A, Martínez MJ et al. Isolated demyelinating syndromes: comparison of different MR imaging criteria to predict conversion to clinically definite multiple sclerosis. Am J Radiology 2000; 21: 702–706.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2008 Issue 2
Most read in this issue
- Multiple sclerosis
- Hemangioblastoma and its treatment using Leksell Gamma Knife
- Treatment Results of Low-Grade Gliomas in Children (a Retrospective Data Analysis)
- Smith-Magenis syndrome: a case report