
Idiopatické generalizované epilepsie s typickými absencemi v dospělosti
Idiopathic generalised epilepsies with typical absences in adults
The syndromes of idiopathic generalised epilepsies (IGEs) manifest with three main types of seizures – typical absences, myoclonic jerks and generalised tonic clonic seizures. Typical absence seizures are frequently underdiagnosed or misinterpreted as focal seizures in adult patients. The correct diagnosis is nevertheless of great practical significance as IGEs demand different treatment to focal epilepsies. Many neurologists still prescribe carbamazepine for typical absences with undesired results – worsening of course of epilepsy (seizures frequency, intensity or developing of absence status). Irrespective of whether all of IGE syndromes constitute a biological continuum or not, knowledge of specific features of each syndrome is required to differentiate them from focal epilepsies. This article details clinical picture of typical absences and special subtypes of absence seizures, e.g. absences with regional myoclonic jerks, and provides characteristics of different IGE syndromes with absences in adult patients.
Key words:
epilepsy - idiopathic generalised epilepsy - absence
Authors:
H. Krijtová; M. Tomášek; P. Marusič
Authors‘ workplace:
Neurologická klinika UK 2. LF a FN Motol
Published in:
Cesk Slov Neurol N 2008; 71/104(1): 34-40
Category:
Review Article
Overview
Typické absence představují spolu s myoklonickými záchvaty a záchvaty generalizovanými tonicko-klonickými trojici záchvatů, jimiž se manifestují idiopatické generalizované epilepsie (IGE). Výskyt typických absencí je u dospělých pacientů lékaři často podceňován a jsou nezřídka zaměňovány za záchvaty fokální. Stanovení správné diagnózy je nicméně zásadní pro volbu vhodného antiepileptika a pro přibližný odhad prognózy. V řadě případů jsou stále pacienti s IGE léčeni karbamazepinem, což má často za následek horší průběh onemocnění – častější či silnější záchvaty, nebo rozvoj epileptického statu. Bez ohledu na to, zda IGE tvoří biologické kontinuum či nikoli, je znalost specifických rysů jednotlivých syndromů nezbytná pro jejich odlišení od fokálních epilepsií. Cílem tohoto sdělení je přehledně připomenout klinický obraz jednotlivých idiopatických generalizovaných epileptických syndromů s absencemi, které se u dospělých pacientů mohou vyskytovat.
Klíčová slova:
epilepsie – idiopatická generalizovaná epilepsie – absence – absence v dospělosti
Úvod
Idiopatické generalizované epilepsie (IGE) jsou onemocnění geneticky podmíněná, věkově vázaná a tvoří 1/3 všech epilepsií. Postihují jinak zdravé jedince obou pohlaví a jsou onemocněním často celoživotním. Typické absence představují spolu s myoklonickými záchvaty a záchvaty generalizovanými tonicko-klonickými (GTCS) trojici záchvatů, jimiž se IGE manifestují. Výskyt typických absencí u dospělých je lékaři často podceňován a chyby při jejich rozpoznávání jsou časté. Přesné stanovení diagnózy je zásadní pro správnou volbu antiepileptika i pro odhad prognózy. Dle různých autorů je asi ¼ až ½ pacientů s IGE vedena pod diagnózou fokální epilepsie a léčena nevhodným antiepileptikem – nejčastěji karbamazepinem [1,2). To má většinou za následek zbytečně horší průběh onemocnění, častější či silnější záchvaty, nebo rozvoj epileptického statu (nejčastěji absencí nebo myoklonického). Cílem tohoto sdělení je podat přehled jednotlivých IGE s absencemi, které se u dospělých pacientů mohou vyskytovat. Základní klinická charakteristika syndromů je vždy doplněna kazuistikou.
Záchvatové projevy
Typické absence jsou definovány jako krátce trvající generalizované záchvaty s náhlým začátkem a náhlým koncem. Mají komponentu klinickou a elektroencefalografickou (EEG). Klinickou komponentu tvoří porucha vědomí různého stupně, která je často doprovázena dalšími symptomy - komponentami. EEG korelátem poruchy vědomí jsou generalizované výboje komplexů hrot-vlna (SW) o frekvenci 3-4 Hz, či jejich varianta – výboje komplexů vícečetných hrotů a pomalé vlny (PSW) [2-4). Atypické absence na rozdíl od typických jsou charakterizovány pozvolným začátkem a pozvolným zakončením, často mají výraznější doprovodné symptomy a iktální EEG je tvořeno pomalými (méně než 2,5 Hz) komplexy SW. Jejich výskyt je spojován s epileptickými encefalopatiemi dětského věku, definice je zde uvedena jen pro odlišení od termínu „typická absence“.
Stupeň poruchy vědomí během typické absence může být různý. Těžká porucha se projeví strnutím, zárazem s prázdným pohledem a areaktivitou. Pacient může udávat „výpadek“, „okénko“, při rozhovoru ztratí kontinuitu. Při středně těžké poruše vědomí pacient může pokračovat v započaté činnosti, ale třeba zpomaleně, s opakováním slov či nesmyslným dokončením věty. Následně může svůj prožitek popisovat jako zúžené vnímání – „vnímání jako přes silné sklo“, „jakoby z jiného světa“, ale také jako tlak, napětí v hlavě, bolest hlavy apod. Lehká porucha vědomí, které si pacient sám vůbec nemusí být vědom, se vyskytuje např. u fantomových absencí. Tento termín byl použit teprve v roce 1993 Fernerem a označuje nenápadné krátké absence, které v běžném životě pacient ani jeho okolí nerozpozná [2, 5-7]. Stupeň poruchy vědomí je závislý na typu syndromu, nejtěžší porucha se vyskytuje u dětské (CAE) nebo juvenilní (JAE) epilepsie s absencemi, lehká či nenápadná porucha se vyskytuje například u juvenilní myoklonické epilepsie (JME). Stupeň poruchy vědomí se může rovněž vyvíjet v průběhu života nebo může u téhož pacienta záchvat od záchvatu kolísat. Absence se mohou vyskytovat spontánně i reflexně, precipitujícím podnětem bývá nejčastěji blikavé světlo, což je v souladu s častým výskytem fotosenzitivity u pacientů s IGE. Porucha vědomí je u absencí většinou provázena dalšími příznaky – komponentami:
Klonická komponenta je tvořena svalovými záškuby, které mohou být rytmické či nepravidelné, izolované či repetitivní. Nejčastěji se vyskytují na začátku absence. Typicky se jedná o záškuby víček, obočí, bulbů, ale i končetin nebo trupu. Tyto záškuby mohou být někdy velmi nenápadné. Nápadné rytmické myoklonie postihující určité regionální svalové skupiny jsou natolik charakteristické, že byly definovány jako zvláštní typy záchvatů (podtypy absencí): myoklonické absence, myoklonie očních víček s absencemi/bez absencí, periorální myoklonie s absencemi/bez absencí. Tyto záchvaty se většinou vyskytují též v rámci některých syndromů, jež svou přítomností definují [2,3].
Tonická komponenta. Tonické záchvaty jako takové se u IGE nevyskytují, ale v průběhu absencí se může objevit tonické napětí některých svalových skupin, nejčastěji v obličeji či na krku, s následnou tonickou změnou polohy, např. retropulzí, nebo i lateropulzí očí a hlavy [2,3].
Automatizmy se u absencí vyskytují běžně, častěji u delších či u absence statu. Liší se od automatizmů při záchvatech parciálních – jsou jednoduché, méně nápadné a nebývají rytmické. Mohou být ale též periorální, nebo pacienti při nich manipulují oděvem či bezcílně bloudí.
Atonická komponenta bývá u dospělých jen naznačena, většinou jako pokles hlavy, brady apod. Pády podmíněné atonií se u dospělých s IGE nevyskytují.
Autonomní komponenta je většinou rovněž málo nápadná, nejčastěji dochází ke zblednutí či zčervenání v obličeji nebo k dilataci zornic. Pacienty udávané pocity v epigastriu mohou připomínat epigastrickou auru temporálních záchvatů.
Byly popsány některé další symptomy, ale ty jsou již vzácnější - pseudoiluze a pseudohalucinace definované jako různé poruchy vnímání (například ztráta prostorového vnímání), jejichž nereálnost si pacient následně uvědomuje, dále fokální motorické projevy apod.
Absence status. Typický absence status epilepticus (ASE) je definován jako protrahovaný, déle než 30 minut trvající nekonvulzivní záchvat s poruchou vědomí doprovázený v EEG generalizovanými SW/PSW komplexy. Porucha vědomí může být i v tomto případě různého stupně a může být provázena dalšími symptomy. Pacient ji popisuje například jako „zpomalené myšlení“, někdy s „mluvením nesmyslů“, nebo pocit „jako v tranzu“, s výpadky částí rozhovoru. Komunikace s okolím vázne, pacienti působí zmateně, jsou většinou zpomalení, dezorientováni v čase, odpovídají jednoduše, nezvládají jednoduché počty, nevybavují si základní údaje. Všechny IGE – snad kromě epilepsie dětského věku s absencemi – se mohou manifestovat ASE. Ten může být buď spontánní nebo provokovaný zevními faktory či nevhodnými léčebnými zásahy [2,3].
EEG nález
Iktální EEG je u typických absencí dle definice tvořeno generalizovanými SW komplexy bilaterálně symetrickými a synchronními 3-4 Hz, které jsou tvořeny hroty a pomalými vlnami s amplitudou kolem 200 μV a s maximem ve střední čáře fronto-centrálně. Nález na EEG však velmi často není zcela symetrický a SW začínají často lateralizovaně, regionálně, se střídavou stranovou převahou. Zakončení může být náhlé nebo několika zpomalujícími se vlnami. Tento vzorec však není typický pro všechny IGE syndromy. Některé se vyznačují výskytem komplexů PSW, nebo kombinací obou typů výbojů. Hranice mezi „iktálními“ a „interiktálními“ EEG výboji není u pacientů s IGE často ostrá a záleží na úrovni a způsobu testování pacienta v průběhu těchto výbojů. Morfologie výbojů může být i značně variabilní, komplexy se mohou objevovat v úsecích kontinuálně či fragmentovaně, jejich frekvence může být pravidelná nebo nepravidelná v rozmezí 3 až 6 Hz. Časté je stranově nekonzistentní maximum a v některých syndromech i vysloveně fokální nálezy hrotů a pomalých vln (nejčastěji s maximem na elektrodách F3 či F4). Tato variabilita a případné fokální nálezy EEG se v současné době vysvětlují hyperexcitabilitou mozkové kůry, která je u IGE nestabilní a může být stranově měnlivá [2,8].
Diagnóza epilepsie je primárně stanovena na základě klinického obrazu, ale popis samotných záchvatů nemusí být pro odlišení generalizovaných epilepsií od fokálních dostačující. Pravděpodobnost záchytu specifické abnormity je u neléčené IGE vysoká, zejména při použití provokačních metod, nicméně v řadě případů může být rutinní EEG nález normální.
Klasifikace IGE syndromů s absencemi u dospělých
V současné době existují dva odlišné názory na klasifikaci IGE. Mezinárodní liga proti epilepsii (ILAE) vytvořila ve své klasifikaci z roku 1989 koncepci jednoho onemocnění vyskytujícího se v adolescenci a v dospělosti a nazvala je IGE s variabilním fenotypem. Toto onemocnění je členěno do 3 podsyndromů podle dominujícího typu záchvatu: juvenilní epilepsie s absencemi, juvenilní myoklonická epilepsie, epilepsie se samotnými GTCS (dříve epilepsie s GTCS po probuzení). Vedle této skupiny onemocnění lze ale podle novějších poznatků předpokládat existenci celé řady dalších samostatných syndromů [4,6]. Ty byly popsány většinou teprve v minulém desetiletí, výjimku tvoří syndrom barvitě popsaný Jeavonsem v roce 1974, a splňují většinu charakteristik IGE, nicméně zatím nebyly do klasifikace ILAE zařazeny [6]. Jednotlivé syndromy se liší věkem začátku a kulminace záchvatů, jejich charakterem a kombinacemi, prognózou a někdy i precipitujícími faktory nebo odpovědí na antiepileptika. Bez ohledu na to, zda jednotlivé IGE syndromy tvoří navzájem biologické kontinuum či nikoli, je znalost specifických rysů jednotlivých syndromů nezbytná pro jejich odlišení od fokálních epilepsií. Přehled a základní charakteristiky IGE, u kterých se vyskytují typické absence a které mohou začínat nebo přetrvávat v dospělosti, uvádí tabulka a podrobně jsou pak jednotlivé syndromy popsány níže.

Juvenilní epilepsie s absencemi (JAE)
Nejnápadnější a nejlépe vyjádřené typické absence se vyskytují u tohoto syndromu. Většina pacientů (80 %) také trpí generalizovanými tonicko klonickými záchvaty a asi 20 % sporadickými myokloniemi [9]. Onemocnění začíná nejčastěji mezi 9.-13. rokem, ale uvádí se až rozmezí mezi 5.-20. rokem věku. GTCS a myoklonie většinou začínají s odstupem 1 roku až 10 let po absencích. Přesná prevalence není známa, ale u dospělých tvoří JAE asi 2-3 % všech epilepsií. Stupeň poruchy vědomí se většinou u jednotlivých pacientů vyvíjí v průběhu onemocnění – na začátku, v dětství, bývá porucha vědomí těžká, zatímco v dospělosti spíše střední. Jemné myoklonie víček iniciálně a nenápadné automatizmy jsou časté. Absence mají podobný charakter jako u CAE, ale často mají delší trvání – nejčastěji 4-30 sekund, někdy trvají i déle. Vyskytují se většinou každodenně, ale s menší četností než u CAE. S diagnózou JAE nejsou slučitelné absence nenápadné a nápadné frekventní myoklonie postihující obličej, nebo končetiny a trup. GTCS se mohou objevit kromě období po probuzení též kdykoliv během dne a též ze spánku. Jsou často rezistentní k terapii. ASE se vyskytuje asi u 1/5 pacientů. Provokujícím faktorem je u absencí rozrušení a hyperventilace, čehož se využívá při standardním EEG vyšetření, u GTCS pak nedostatek spánku, alkohol, únava a méně často blikavé světlo. EEG interiktální může být i normální, nebo lze někdy zachytit asymetrické výboje SW/PSW nízké amplitudy.
Odlišení od CAE může být zpočátku obtížné, nicméně výskyt GTCS a myoklonií současně s absencemi doprovázenými těžkou poruchou vědomí svědčí pro JAE. Oproti CAE také většinou začínají absence v pozdějším věku a jsou méně frekventní. U Jeavonsova syndromu jsou absence výrazně kratší s nápadným rytmickým mrkáním a u pacientů s PMA dominují periorální myoklonie. Odlišení od JME by nemělo být složité, protože se jedná o syndrom s dominujícím jiným typem záchvatů. K záměně typických absencí za parciální komplexní záchvaty dochází u dospělých mnohem častěji než u dětí. Tomu napomáhá delší trvání absencí u dospělých, častý výskyt automatizmů a neúplná porucha vědomí s různě změněným a pacienty často bizarně líčeným vnímáním okolí. Nakupení absencí následované generalizovaným konvulzívním záchvatem může rovněž být považováno za komplexní parciální záchvat se sekundární generalizací.
Kazuistika
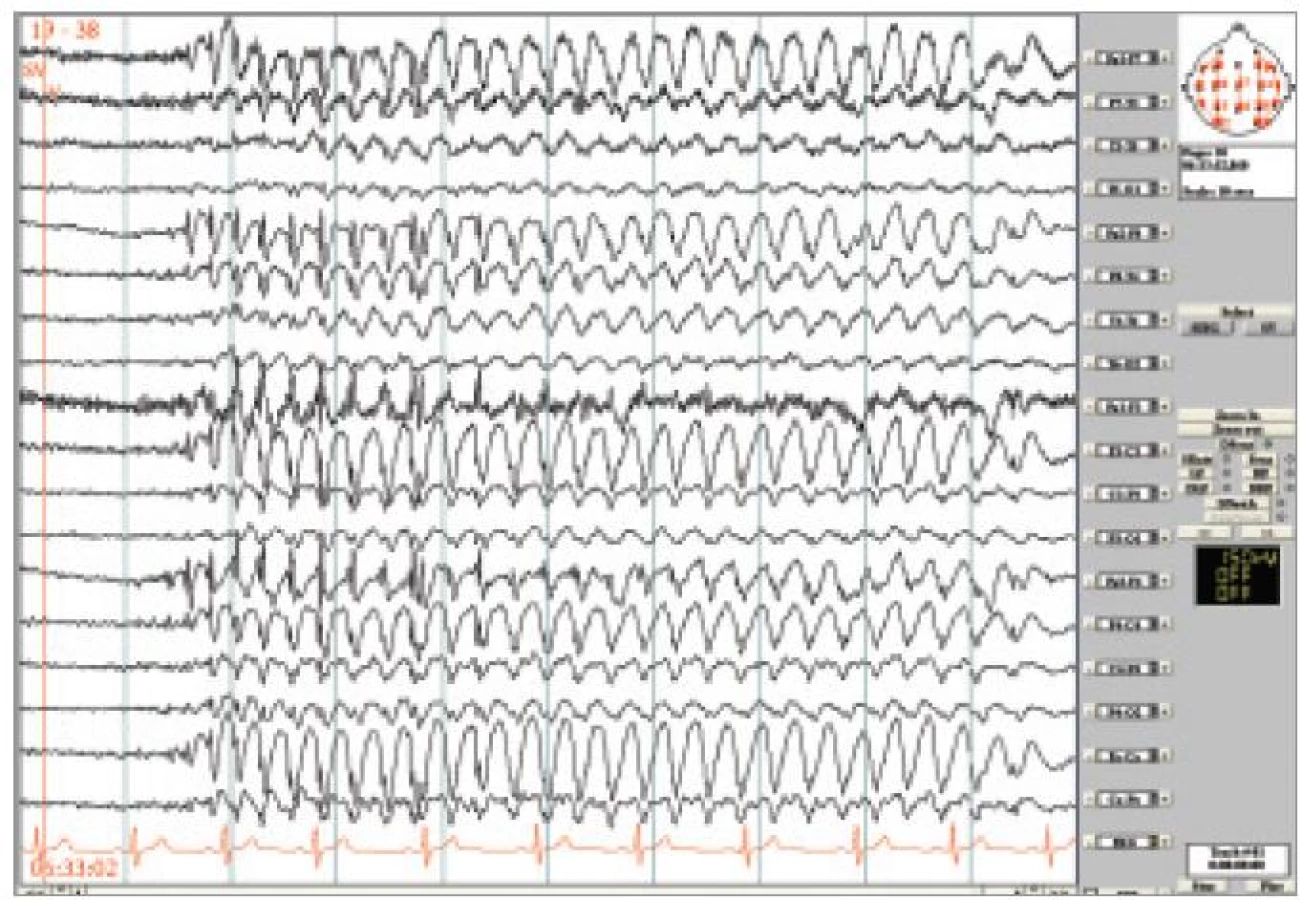
23letá žena byla odeslána k video-EEG monitorování s diagnózou epilepsie se záchvaty grand mal atypicky reagující na podávání antiepileptik, ke zvážení epileptochirurgického zákroku. V rodinné anamnéze byl údaj o výskytu záchvatů u sestry matky. Nemocná prodělala v 7 letech úraz hlavy s kontuzí mozku a frakturou zygomatického oblouku. Záchvaty se začaly objevovat asi rok po úrazu, tedy v 8 letech, měly charakter zárazu s výpadkem vědomí, frekvence asi 1-2/den, a objevovaly se i přes léčbu v období od 8 do 14 let. Asi od 15 let mívala 1/rok GTCS a malé záchvaty se přestaly vyskytovat. V posledních 2 letech se přidaly nepříjemné stavy trvající cca 10-15 minut – vnímala okolí jako po své smrti, někdy s úzkostí, nauzeou, jakoby ve vlnách. Tyto stavy byly několikrát zakončeny GTCS, ale většinou spontánně odezněly a vyskytovaly se cca 1/měsíc. Záchvaty se provokovaly nedostatkem spánku, stresem nebo námahou. Žena byla léčena nejprve karbamazepinem (CBZ), poté v adolescenci valproátem (VPA) a poslední 2 roky lamotriginem (LTG) s maximální dávkou 250 mg/den. EEG bylo dle dokumentace v minulosti s nálezem pro epilepsii specifickým, blíže neupřesněným, ale v poslední době bez abnormit. MRI mozku bylo bez patologických změn. Při video-EEG jsme zaznamenali četné generalizované výboje SW 3-3,5 Hz v trvání až 15 sekund (obr. 1). Pokud se výboje objevily při rozhovoru, ztratila pacientka nit a bezradně dokončila větu do neurčita. Po skončení výbojů ihned správně navázala a pokračovala ve vyprávění. Po upozornění nemocné, že se jednalo o záchvat si uvědomila, že takové problémy mívá, ale netušila, že jsou to také záchvaty. U této nemocné tedy došlo v průběhu dospívání k typickému zmírnění stupně poruchy vědomí během absencí. Protrahované stavy s poruchou vnímání („jako po své smrti“) mohou odpovídat delším absencím, podobné jsou popisovány u absence statu.
Juvenilní myoklonická epilepsie (JME)
Typické absence se vyskytují asi u 1/3 nemocných s JME [10]. Většinou v historii onemocnění předcházejí myokloniím. Jsou kratšího trvání a méně nápadné než u pacientů s CAE nebo JAE, jsou málo frekventní a někdy udávané až na cílený dotaz, neboť nejsou v popředí pozornosti pacientů a jejich okolí. Podrobnější údaje o JME nejsou předmětem tohoto sdělení.
IGE s fantomovými absencemi
Tento syndrom je charakterizovaný následující klinickou trias:
- fantomové absence
- nečetné neprovokované GTCS v dospělosti – většinou hodnocené jako první klinická manifestace epilepsie
- ASE – asi u 50 % pacientů
Předpokládá se prevalence kolem 10 % všech IGE [7]. Fantomová absence je definována jako nenápadná krátce trvající porucha vědomí s EEG korelátem nepravidelných generalizovaných SW 3-6 Hz trvajících 2-5 sekund. V běžném životě si pacient tuto poruchu neuvědomuje, ale lze ji prokázat při testování pozornosti či reaktivity. Fantomové absence se vyskytují nepochybně dlouho před prvním neprovokovaným GTCS, jenž se většinou objeví až v dospělosti. Tomuto GTCS někdy předchází ASE. Onemocnění má patrně genetický podklad a trvá po celý život [2,5-7]. Omyly při stanovení diagnózy u pacientů s tímto syndromem jsou časté. Většina neurologů vyhodnotí první neprovokovaný GTCS buď jako ojedinělý záchvat, nebo jako projev fokální epilepsie. Porucha vědomí při absence statu předcházejícím GTCS je v takovém případě považována za důkaz pro fokální začátek záchvatu, čemuž dále napomáhají časté fokální abnormity v EEG. Dříve nebo později se ale v EEG zachytí bilaterální krátké generalizované výboje SW v trvání do 5 sekund.
Kazuistika
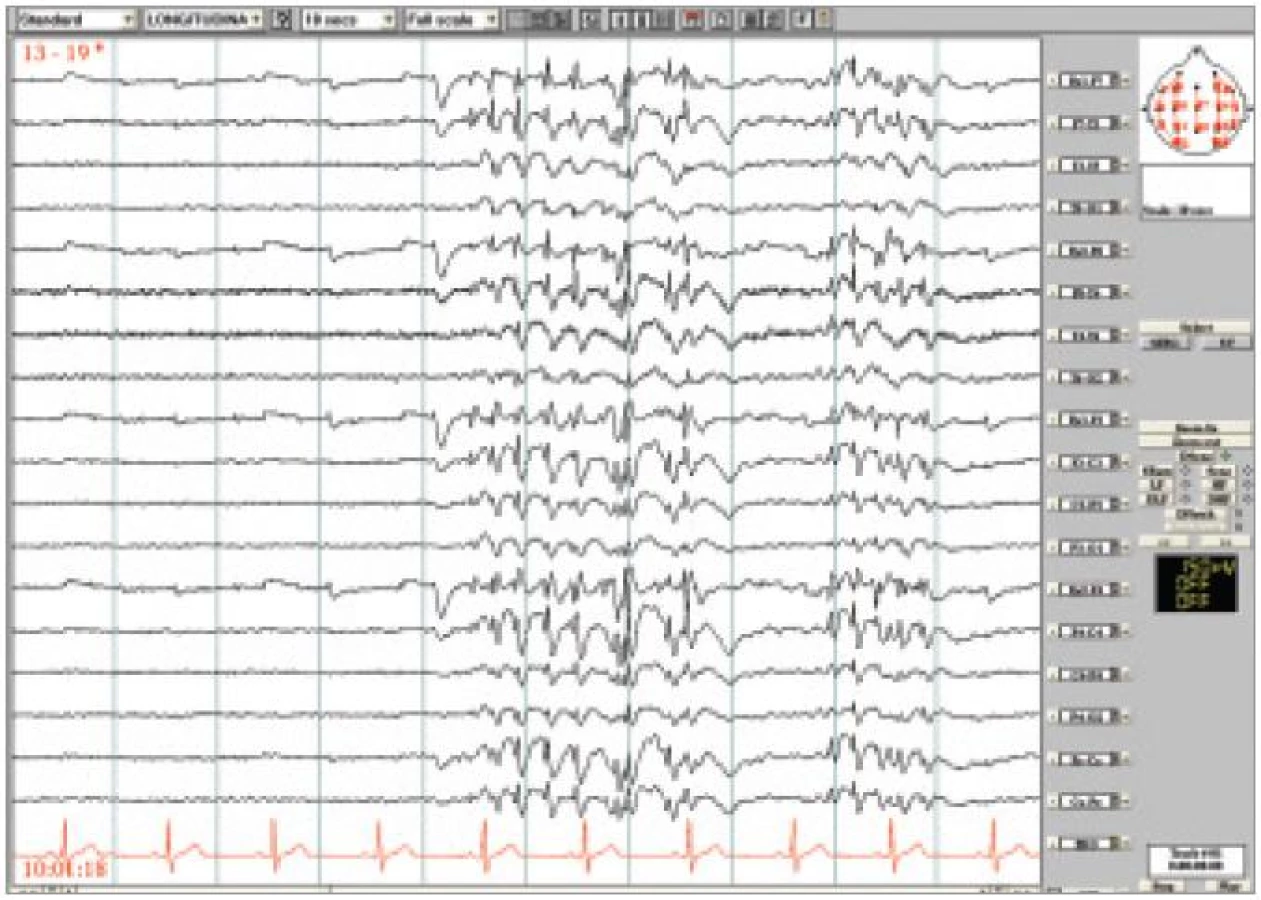
26letý muž byl odeslán k video-EEG monitorování s diagnózou stp. 1. neprovokovaném GTCS, MRI nelezionální, v EEG generalizované SW výboje. K posouzení kognitivní poruchy během SW výbojů. Byly zachyceny četné úseky generalizovaných výbojů SW 3-6 Hz, které trvaly až 5 sekund. Objevovaly se většinou při ospalosti, často po zavření očí, ale i při očích otevřených. Převážně neměly žádný behaviorální korelát, jen ojediněle bylo možno pozorovat mrkání nebo lehký pokles zvednutých horních končetin. Nejnápadnější změny byly zachyceny v průběhu vyťukávání textové zprávy na mobilním telefonu, kdy současně se začátkem výbojů (obr. 2) došlo k zárazu v této činnosti, ale v pauze mezi výboji a po jejich skončení vyťukávání pokračovalo.
Periorální myoklonie s absencemi (PMA)
Typické absence s periorálními myokloniemi jako dominujícím typem záchvatu spolu s dalšími symptomy konstituují IGE syndrom, který rovněž zatím nebyl zařazen do ILAE klasifikace [6,11-13]. Věk postižených v době začátku záchvatů se pohybuje mezi 2-13 roky, děvčata jsou o něco častěji postižena než chlapci. V dospělosti pak tento syndrom tvoří asi 9 % případů epilepsie s typickými absencemi. PMA jsou charakterizovány záškuby periorálního svalstva, které se mohou šířit i na končetiny. Současně se může dostavit porucha vědomí, která bývá různého stupně. Záškuby m. orbicularis oris vedou k protruzi rtů, záškuby m. depressor anguli oris stahují koutky dolů nebo nejméně často se jedná o záškuby žvýkacích svalů, což může být provázeno cvakáním čelistí. Pacienti si jsou většinou těchto záškubů vědomi, mohou být ojedinělé nebo rytmické a šíření na končetiny může napodobovat jacksonský rozvoj u fokálních záchvatů. Frekvence záchvatů je většinou vysoká. GTCS se vyskytují u všech pacientů, často jsou uváděny nakupením PMA nebo ASE. V EEG se interiktálně často objevují abortivní generalizované SW/PSW 4-7 Hz, časté jsou asymetrie i stranové střídání abnormity. Není přítomna fotosenzitivita. Ve většině případů je toto onemocnění refrakterní k terapii.
Samotný údaj o výskytu periorálních myoklonií nelze ještě považovat za dostatečný pro stanovení diagnózy PMA, neboť se tento typ záchvatů může vyskytovat i u jiných IGE (spontánně nebo reflexně) nebo u atypických absencí v rámci symptomatických epilepsií. Zde ale periorální myoklonie nejsou nejnápadnějším a převažujícím typem záchvatů.
PMA je často zaměněna za fokální epilepsii kvůli záškubům vnímaných pacienty i jejich okolím jednostranně a kvůli fokálním projevům v EEG. Odlišení od JAE nebývá složité, neboť myoklonické záškuby jsou u PMA většinou nápadnější než porucha vědomí.
Kazuistika
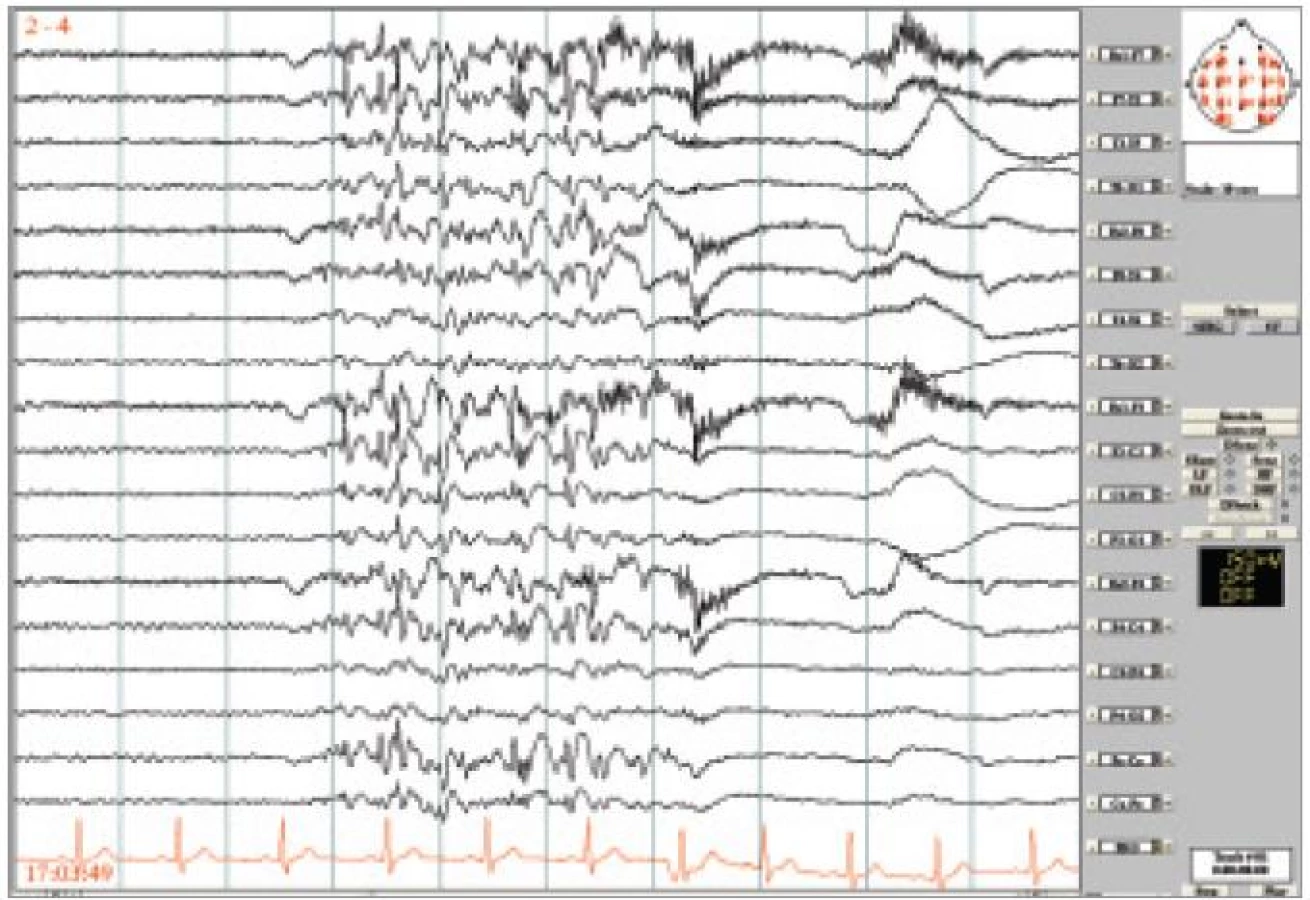
36letý muž byl odeslán k video-EEG monitorování s diagnózou fokální epilepsie, záchvaty simplexní motorické (klonické levostranné s jacksonským rozvojem) a sekundární GTCS. MRI nelezionální, etiologie neznámá, farmakorezistentní. Ke zvážení epileptochirurgického výkonu. Záchvaty se začaly vyskytovat ve 4 letech věku – jednalo se o záškuby levého koutku (dříve uváděno i pravého), izolované či repetitivní, trvající několik sekund – i déle, někdy s šířením na LHK a LDK, či bilaterálně s trváním až desítky minut a s částečně zastřeným vědomím. GTCS měl asi 4× za život. U pacienta byla vyzkoušena všechna dostupná antiepileptika (kromě gabapentinu) v různých kombinacích. Záchvaty pozoroval asi 4 za den. Během monitorování byly zachyceny četné záchvaty s dominujícími záškuby m. depressor anguli oris bilaterálně, někdy iniciálně s jemnými sotva patrnými myokloniemi víček a následně záškubem ramen. V EEG byly záškuby provázeny generalizovanými nepravidelnými výboji SW/PSW se střídavou stranovou převahou (obr. 3).
Jeavonsův syndrom (myoklonie očních víček s absencemi, EMA)
Tento syndrom se vyznačuje výskytem zvláštního typu záchvatů – myoklonií očních víček, které mohou, ale nemusejí být provázeny lehkým zúžením vědomí. Záškuby očních víček jsou poměrně nápadné a současně lze pozorovat stočení bulbů vzhůru. Kromě tohoto typu záchvatů je Jeavonsův syndrom dále charakterizován fotosenzitivitou a typickou provokací záchvatů a/nebo EEG výbojů zavřením očí. Z tohoto důvodu by měl být podle některých autorů tento syndrom zařazen mezi reflexní generalizované epilepsie. GTCS se vyskytují jen zřídka, ale u většiny pacientů. Záchvaty začínají v dětství, nejčastěji mezi 6.-8. rokem, a přetrvávají do dospělosti. Prevalence je uváděna asi 3 % u dospělých pacientů s epilepsií [14]. Častěji jsou postiženy ženy. Záchvaty EMA mohou být součástí nejen idiopatických epileptických syndromů. V nedávné době byly popsány záchvaty téhož charakteru u pacientů s epilepsií symptomatickou [15]. Tito pacienti se vyznačují anamnézou perinatální zátěže, lehkou mentální retardací, lehce zpomalenou základní aktivitou v EEG a někdy i bilaterálními změnami na MRI [16,17]. Nicméně v těchto případech není většinou vyjádřena fotosenzitivita a záchvaty se vyskytují častěji i nezávisle na zavření očí.
Kazuistika
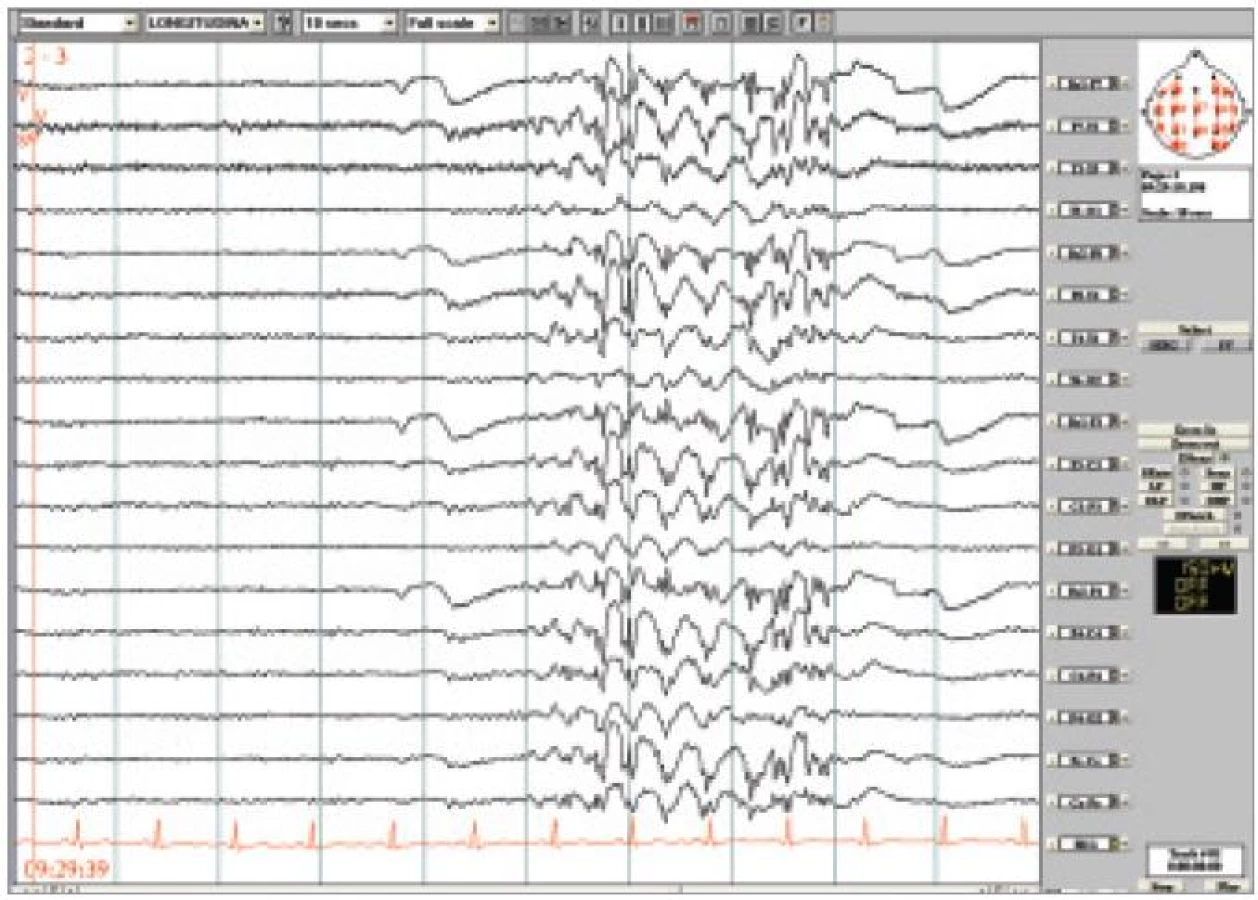
43letá žena byla doporučena k video-EEG monitorování k posouzení kognitivní poruchy během výbojů SW/PSW komplexů, které byly náhodně zachyceny při EEG vyšetření indikovaném z jiného důvodu. MRI bylo bez patologických změn. Na cílené dotazy připustila nemocná kratičké stavy se zakoukáním a mrkáním víček, které se u ní objevovaly od dětství, většinou při únavě, a které považovala za oční poruchu. Nikdy neprodělala GTCS. Při video-EEG byly zachyceny četné záchvaty se stočením bulbů vzhůru a prudkým frekventním mrkáním. V EEG tyto záchvaty byly provázeny nepravidelnými výboji SW/PSW až 5 sekund dlouhými (obr. 4). Při testování reaktivity bylo během těchto výbojů prokázáno prodloužení reakční doby.
Závěr
Výskyt typických absencí není pouze záležitostí dětského věku. Běžně se vyskytují i v dospělosti – u 9 % pacientů s epilepsií – a jsou v některých případech refrakterní na léčbu. Stupeň poruchy vědomí u absencí může být různý v závislosti na syndromu IGE a může se u jednoho pacienta s věkem změnit, většinou zmírnit, ale může i kolísat záchvat od záchvatu. Některé IGE se manifestují až v dospělém věku a stanovení správné diagnózy může být obtížné. Asymetrický nález v EEG, asymetrický projev motorický a zavádějící nebo neurčitý popis klinických projevů mohou vést k nesprávnému závěru, který s sebou nese riziko nesprávně zvolené léčby. Při diagnostických pochybnostech je vhodné volit k léčbě některé ze „širokospektrých“ antiepileptik a/nebo využít možnosti upřesnění diagnózy při video-EEG monitorování.
Přijato k recenzi: 1. 3. 2007
Přijato do tisku: 4. 9. 2007
Korespondující autor:
MUDr. Hana Krijtová
Neurologická klinika UK 2. LF a FN Motol
V Úvalu 84
150 06 Praha 5
E- mail: hana.krijtova@fnmotol.cz
Sources
1. Benbadis SR, Tatum WO, Gieron M. Idiopathic generalized epilepsy and choice of antiepileptic drugs. Neurology 2003; 61(12): 1793-5.
2. Panayiotopoulos CP. Idiopathic Generalised Epilepsies. In: Panayiotopoulos CP, editor. The Epilepsies: Seizures, Syndromes and Management. Chipping Norton: Bladon Medical Publishing 2005: 271-339.
3. Duron RM, Medina MT, Martinez-Juarez IE, Bailey JN, Perez-Gosiengfiao KT, Ramos-Ramirez R et al. Seizures of idiopathic generalized epilepsies. Idiopathic generalized epilepsies recognized by the International League Against Epilepsy. Use and abuse of EEG in the diagnosis of idiopathic generalized epilepsies. Epilepsia 2005; 46(Suppl 9): 34-47.
4. Nordli DR, Jr. Idiopathic generalized epilepsies recognized by the International League Against Epilepsy. Epilepsia 2005; 46(Suppl 9): 48-56.
5. Ferner RE, Panayiotopoulos CP. 'Phantom' typical absences, absence status and experiential phenomena. Seizure 1993; 2(3): 253-256.
6. Panayiotopoulos CP. Syndromes of idiopathic generalized epilepsies not recognized by the International League Against Epilepsy. Epilepsia 2005; 46(Suppl 9): 57-66.
7. Panayiotopoulos CP, Koutroumanidis M, Giannakodimos S, Agathonikou A. Idiopathic generalised epilepsy in adults manifested by phantom absences, generalised tonic-clonic seizures, and frequent absence status. J Neurol Neurosurg Psychiatry 1997; 63(5): 622-627.
8. Koutroumanidis M, Smith S. Use and abuse of EEG in the diagnosis of idiopathic generalized epilepsies. Epilepsia 2005; 46(Suppl 9): 96-107.
9. Panayiotopoulos CP. Absence epilepsies. In: Engel J, Pedley TA, editors. Epilepsy: a comprehensive textbook. Philadelphia: Lippincott-Raven 1998: 2327-2346.
10. Panayiotopoulos CP, Obeid T, Waheed G. Absences in juvenile myoclonic epilepsy: a clinical and video-electroencephalographic study. Ann Neurol 1989; 25(4): 391-397.
11. Baykan B, Noachtar S. Perioral myoclonia with absences: an overlooked and misdiagnosed generalized seizure type. Epilepsy Behav 2005; 6(3): 460-462.
12. Bilgic B, Baykan B, Gurses C, Gokyigit A. Perioral myoclonia with absence seizures: a rare epileptic syndrome. Epileptic Disord 2001; 3(1): 23-27.
13. Clemens B. Perioral myoclonia with absences? A case report with EEG and voltage mapping analysis. Brain Dev 1997; 19(5): 353-358.
14. Giannakodimos S, Panayiotopoulos CP. Eyelid myoclonia with absences in adults: a clinical and video-EEG study. Epilepsia 1996; 37(1): 36-44.
15. Mayer TA, Schroeder F, May TW, Wolf PT. Perioral reflex myoclonias: a controlled study in patients with JME and focal epilepsies. Epilepsia 2006; 47(6): 1059-1067.
16. Scuderi C, Musumeci SA, Ferri R, Calabrese G, Elia M. Eyelid myoclonia with absences in three subjects with mental retardation. Neurol Sci 2000; 21(4): 247-250.
17. Sevgi Demirci EB, Saygi S. Unusual features in eyelid myoclonia with absences: a patient with mild mental retardation and background slowing on electroencephalography. Epilepsy Behav 2006; 8(2): 442-445.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2008 Issue 1
Most read in this issue
- Bezpečnosť MRI vyšetrenia u pacientov s kovovými implantátmi a implantovanými prístrojmi
- Difuzní gliomy mozkového kmene u dětí. Noční můra dětského onkologa.
- Myasténia gravis
- Klinické využití protilátek u roztroušené sklerózy