
Miller Fisherův syndrom – čtyři vlastní pozorování a přehled současných poznatků
Miller Fisher Syndrome – Four Case Reports and Review of Current Concept
Miller Fisher syndrome (MFS) is a rare disorder considered to be a clinical variant of Guillain-Barré syndrome (GBS), an acute post-infection inflammatory disorder of the peripheral nerves. The classic triad of ophthalmoplegia, ataxia and areflexia is often clinically recognizable, but sometimes other cranial nerves may be affected, especially the caudal (bulbar syndrome) or facial nerves. MFS is generally regarded as a self-limiting, benign condition with a good prognosis and recovery with no residual deficits. Overlap syndromes sharing features of GBS with weakness of limb and even respiratory muscles may occur rarely. The anti-ganglioside antibodies anti-GQ1b IgG are found in more than 90% of MFS patients and are an excellent diagnostic marker. In this review, we report four cases of MFS confirmed by the presence of anti-GQ1b antibodies and summarize characteristics of its neurological symptoms and signs, the current state of knowledge about pathogenesis, and findings relevant to correct diagnosis. Related disorders, which may be accompanied by positive anti-GQ1b antibodies, are also mentioned.
Key words:
Miller Fisher syndrome – antiganglioside antibodies – molecular mimicry – anti-GQ1b syndrome
Autoři:
Z. Ambler; J. Valeš
Působiště autorů:
Neurologická klinika LF UK a FN Plzeň
Vyšlo v časopise:
Cesk Slov Neurol N 2011; 74/107(6): 689-694
Kategorie:
Kazuistika
Souhrn
Miller Fisherův syndrom (MFS) je vzácná porucha a považuje se za samostatnou variantu Guillainova-Barrého syndromu (GBS), akutní postinfekční zánětlivou poruchu periferních nervů. Je charakterizován akutním začátkem trias oftalmoplegie, ataxie a areflexie, ale někdy mohou být postiženy i další hlavové nervy, především kaudální (bulbární syndrom) nebo n. facialis. MFS je benigní onemocnění s dobrou prognózou a úpravou bez reziduálního deficitu. Jen vzácně může dojít k progresi do smíšené formy s klasickým GBS se slabostí končetinového, ale i dýchacího svalstva. Antigangliosidové protilátky anti-GQ1b IgG bývají přítomny u více než 90 % pacientů s MFS a představují základní diagnostický marker. V práci popisujeme čtyři vlastní případy potvrzené přítomností anti-GQ1b protilátek a shrnujeme klinický obraz, současné názory na patogenezi, nálezy významné pro diagnózu, ale i příbuzné jednotky, které mohou být doprovázeny pozitivitou anti-GQ1b protilátek.
Klíčová slova:
Miller Fisherův syndrom – antigangliosidové protilátky – molekulární mimikry – anti-GQ1b syndrom
Úvod
Miller Fisherův (MFS) je vzácná klinická varianta Guillainova-Barrého syndromu (GBS). Jedná se o akutní postinfekční, imunitně zprostředkovanou polyneuropatii s charakteristickou klinickou trias – oftalmoplegie, ataxie a areflexie, kterou jako první popsal bostonský neurolog Charles Miller Fisher v roce 1956 [1]. Současně se však mohou objevovat i další příznaky z postižení hlavových nervů, především kaudálních. Vyjma klinického obrazu je v diagnostice přínosné vyšetření protilátek proti gangliosidům v séru, vyšetření mozkomíšního moku, elektromyografie (EMG) a magnetická rezonance (MR) mozku. Příznaky většinou vznikají během několika dní a odezní spontánně nebo po léčbě za několik týdnů. Názory na léčbu nejsou jednotné, ale prognóza je většinou dobrá. Vzhledem k tomu, že onemocnění je vzácné, klinická symptomatika do určité míry variabilní a diagnóza není vždy jednoduchá, uvádíme čtyři vlastní případy MFS.
Soubor
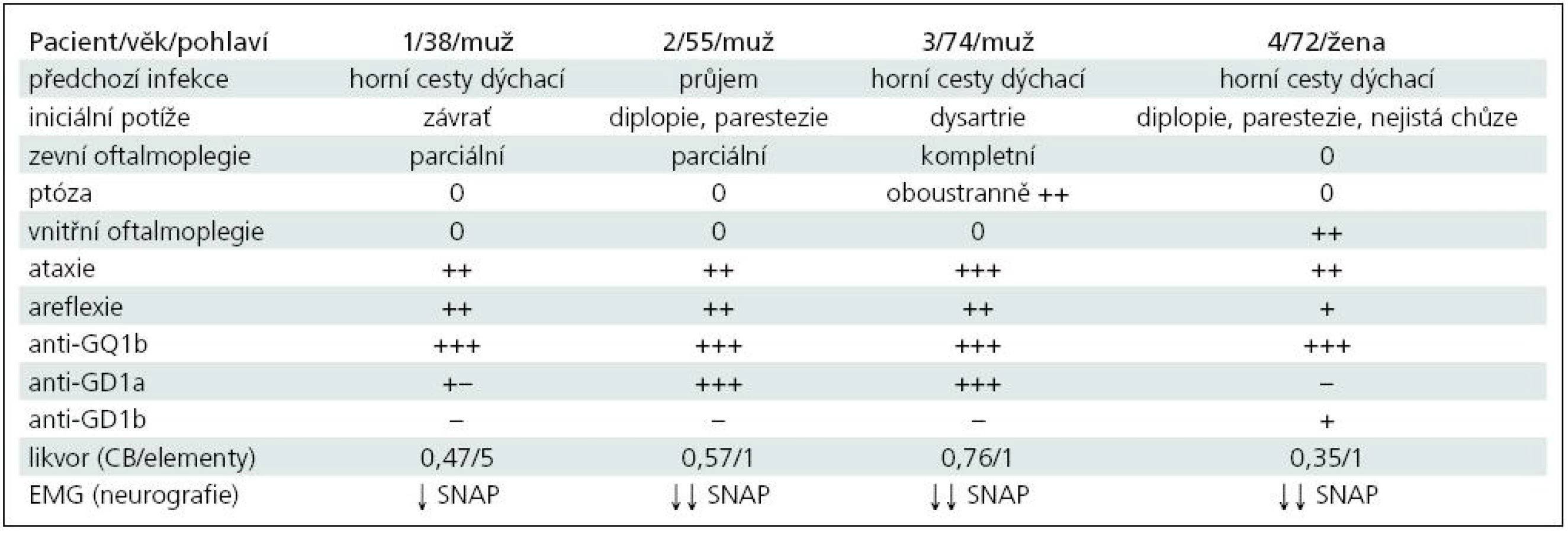
Kazuistika 1
Muž 39 let, v anamnéze 1× ledvinná kolika.
Asi v polovině ledna začaly bolesti v krku a teploty přes 38 °C. Po léčbě perorálními antibiotiky se stav zlepšil, ale po jejich dobrání došlo k recidivě potíží. Byl proto znovu léčen perorálními antibiotiky, opět jen s přechodným efektem. Koncem února následovala další recidiva, kdy byl již léčen i.v. antibiotiky, ale i nadále přetrvávaly subfebrilie a bolesti v krku. 7. 3. se objevila závrať, tlak za očima, další den brnění prstů obou rukou. 10. 3. byl přijat na Neurologickou kliniku FN Plzeň (NK FN). Při přijetí byl lucidní, subfebrilní, bez poruchy řeči, oční bulby nedotahoval do krajních poloh vertikálně i horizontálně oboustranně, zornice normálně reagovaly na fotoreakci i akomodaci. Na horních a dolních končetinách (HK, DK) byla normální hybnost i síla, šlachookosticové reflexy (ŠOR) na pravé HK byly výbavné jen C5/6, na levé HK ŠOR C5/8 byly velmi nízké, na DK byla areflexie L2/S2. Na HK i DK byla výrazná ataxie. Akrálně na HK i DK byla mírná taktilní a algická hypestezie, polohocit i ladička byly normální. Sed byl možný jen s oporou, ve stoji II a III byly výrazné titubace a chůze byla nejistá o široké bázi.
V mozkomíšním moku byla celková bílkovina 0,47 g/l, mononukleáry 5/ul, glukóza 3,3 mmol/l, laktát 1,5 mmol/l. Byly prokázány dva oligoklonální pásy bez sérového korelátu, jeden ve slabě alkalické a jeden v neutrální oblasti. Na MR mozku byl normální nález.
Při EMG neurografii byly zjištěny nízké amplitudy senzitivních nervových akčních potenciálů (SNAP) ortodromně na n. ulnaris (1,4 µV) a n. medianus (1,6 µV), na DK normální, sumační svalové akční potenciály (CMAP) na HK i DK i ostatní parametry kondukční studie byly normální.
Protilátky proti gangliosidům: anti-GQ1b 200,2 %; anti-GD1a 29,9 %; anti-asialo-GM1 15,1 %; anti-GM2 10,2 % (laboratoř Interimun Pardubice – interpretace: hodnoty < 30 % negativní, 30–50 % hraniční, 50–100 % pozitivní, > 100 % silně pozitivní).
Nemocný byl léčen intravenózním imunoglobulinem (IVIG) v celkové dávce 160 g (2 g/kg). IVIG byl podán až druhý den po přijetí, kdy se rozvinula bulbární symptomatika. Přesto došlo k další progresi, byla přítomna chabá dysartrie, dysfonie, rinofonie, paradoxní dysfagie s nutností výživy nazogastrickou sondou. Zlepšování začalo 10. den po přijetí, po 13 dnech hospitalizace se upravila řeč i polykání, trvala lehká porucha hybnosti očních bulbů, byl schopen samostatné chůze s lehkou oporou. Při kontrole za pět měsíců přetrvávalo brnění konečků prstů všech končetin, areflexie L5/S2, ostatní nález byl normální. Kontrolní neurografie prokázala výrazné zlepšení amplitud SNAP (n. ulnaris 6,1; n. medianus 12 µV).
Kazuistika 2
Muž 55 let, v anamnéze byla půl roku léčena arteriální hypertenze, vrozená koktavost a obezita. Kouří 10 cigaret denně a udával 3–5 piv denně.
V červenci byl v Mexiku, následně již doma měl několik dní lehký průjem bez teplot. 9. 8. se objevilo dvojité vidění a brnění aker končetin oboustranně. Od dalšího dne začal huhňat, zhoršovalo se polykání, zakuckával se, tekutiny se mu vracely nosem a objevila se i nejistota při chůzi.
Byl přijat nejprve na spádové interní oddělení a pro progresi potíží po třech dnech přeložen na neurologii s diagnózou suspektní porucha nervosvalového přenosu, dif. dg. psychogenní původ potíží. 13. 8. vzhledem k další progresi polykacích potíží přeložen na NK FN.
Při přijetí byl lucidní, řeč byla huhňavá, měl lehkou dysfagii při polykání tekutin, výrazně omezený pohyb bulbů do všech stran, obě zornice normálně reagovaly, patrové oblouky byly oboustranně pokleslé. Na HK i DK byla normální hybnost i síla, ŠOR byly oboustranně výbavné jen C5/6, C7/8 a L2/S2 byla oboustranně areflexie, čití bylo normální. Chůze byla ataktická o široké bázi. V mozkomíšním moku byla celková bílkovina 0,57 g/l, mononukleár 1/ul, glukóza 3,8 mmol/l, laktát 1,9 mmol/l.
Neurografie prokázala nízké amplitudy SNAP na HK (n. ulnaris 1,8; n. medianus 1,2; n. radialis 5,9 µV), na DK byly SNAP nevýbavné. Na n. ulnaris a peroneus byly hraniční latence F odpovědí.
Protilátky proti gangliosidům: anti-GQ1b 173,6 %; anti-GD1a 148,2 %; anti--asialo-GM1 15,1 %.
Nemocný byl léčen IVIG v celkové dávce 230 g (2 g/kg). Již následující den po IVIG se zlepšila řeč, dysfagie, stále však přetrvávalo výrazné omezení pohybu obou očních bulbů všemi směry, téměř vymizely parestezie. Byl propuštěn 19. 8. po sedmi dnech hospitalizace, kdy byla i výrazně zlepšena chůze. Při kontrole za měsíc byl již pohyb očními bulby normální, trvala areflexie L5/S2 oboustranně a mírné parestezie na dlaních a ploskách, stoj a chůze byly normální.
Kazuistika 3
Muž 74 let, diabetik 2. typu léčený perorálními antidiabetiky a inzulinem, léčil se rovněž pro arteriální hypertenzi.
Začátkem dubna měl virózu, od 23. 4. se začala horšit řeč, huhňal, 24. 4. mu pokleslo pravé a další den levé oční víčko. 26. 4. se objevila diplopie, brnění prstů HK a DK oboustranně a zhoršení stability při chůzi. Od 24. 4. byl hospitalizován na interním oddělení, 30. 4. přeložen na NK FN.
Při přijetí byl lucidní, měl dysartrii, výraznou ptózu oboustranně, nebyl možný pohyb očními bulby žádným směrem, obě zornice normálně reagovaly. Na HK i DK byla normální hybnost i síla, ŠOR C5/7 oboustranně byly nízké, C8 i L2/S2 byla areflexie. Na HK byla lehce nepřesná taxe, na DK výrazná ataxie. Povrchová citlivost byla normální, pallanestezie od kolen distálně. Chůze byla výrazně ataktická s tendencí k pádu. V mozkomíšním moku byla celková bílkovina 0,76 g/l, mononukleár 1/ul, glukóza 6,0 mmol/l, laktát lehce zvýšen na 2,6 mmol/l (norma 1,2–2,1).
Při neurografii byly výrazně snížené amplitudy nebo nevýbavné SNAP na HK (n. ulnaris 0; n. medianus 0,6; n. radialis 1,3 µV), nevýbavné na DK. Na DK byla mírná časová disperze CMAP a prodloužené minimální latence F vln (F-M na n. tibialis 58 ms – 3,4 SD, na n. peroneus 60 ms – 4,9 SD). Neurografický nález mohl být ovlivněn i současnou diabetickou neuropatií.
Protilátky proti gangliosidům: anti-GQ1b 157,2 %; anti-GD1a 156,7 %. Ostatní v normě.
Byl léčen IVIG v celkové dávce 170 g (2 g/kg). Po léčbě došlo k postupnému zlepšování neurologických potíží. Stav byl ale komplikován atrio-ventrikulární blokádou II.–III. stupně a bylo indikováno zavedení trvalé kardiostimulace. Poruchy srdečního rytmu nejsou popisovány jako nežádoucí účinek IVIG, a proto shodně s kardiology usuzujeme, že šlo spíše o koincidenci bez kauzálního vztahu k MFS nebo terapii. Pacient byl přeložen na interní oddělení osmý den hospitalizace s přetrvávající výraznou zevní oftalmoplegií (pohyb do všech směrů 1–2 mm), ale s jen mírnou ataxií při chůzi. Na plánovanou kontrolu se nedostavil.
Kazuistika 4
Žena 72 let s léčenou arteriální hypertenzí.
V polovině ledna měla virózu bez teplot, měla jen rýmu a kašel a byla unavená. Léčena perorálními antibiotiky. Od 28. 1. se objevila nejistota při chůzi, dvojité vidění pří pohledu doleva, brnění obou rukou a na chodidlech. Při přijetí na NK FN 29. 1. byla lucidní, afebrilní, oční bulby ve středním postavení, volně pohyblivé všemi směry, bez nystagmu, udávala diplopii při pohledu dopředu a nahoru. Zornice byly izokorické, mydriatické s jen naznačenou fotoreakcí. ŠOR C5/8 byly normálně výbavné, L2/4 jen ve stopě po facilitaci, areflexie L52/S2. Na HK byla oboustranně lehká ataxie, výraznější ataxie na DK, stoj II a III byl nejistý, chůze o širší bázi a výrazně ataktická.
V mozkomíšním moku byla celková bílkovina 0,35 g/l, mononukleár 1/ul, glukóza 6,0 mmol/l, laktát lehce zvýšen na 2,6 mmol/l.
Při neurografii byly nevýbavné SNAP na HK a n. suralis, nízká amplituda na n. peroneus (4,3 µV).
Protilátky proti gangliosidům: anti-GQ1b 163 %; anti-GD1b 63%.
Byla léčena IVIG v celkové dávce 160 g (2 g/kg) a stav se postupně zlepšoval. Při kontrole v dubnu ustoupily parestezie i diplopie, chůze byla již jistá, bulby byly volně pohyblivé všemi směry, stále byly mydriatické zornice s jen naznačenou fotoreakcí, lehce nepřesná taxe, hyporeflexie L2/4 a areflexie L5/S2.
Výsledky
U všech našich nemocných šlo o MFS (tab. 1). Diagnóza byla stanovena na základě klinické symptomatiky, EMG (neurografie) a ve všech případech ji potvrdila výrazná přítomnost protilátek anti-GQ1b. U 1., 2. a 3. pacienta kromě ataxie a areflexie byla přítomna jen zevní oftalmoplegie (pouze u 2. současně i ptóza) a byl i různě vyjádřen bulbární syndrom. Kromě protilátek anti-GQ1b byly přítomny i anti-GD1a, které se vyskytují hlavně u axonální varianty GBS. U 4. pacientky byla přítomna pouze vnitřní oftalmoplegie (kromě diplopie), která se jako samostatná vyskytuje u MFS velmi vzácně. Byly také lehce zvýšeny protilátky anti-GD1b, jež se častěji vyskytují s ataktických forem GBS. Vyšetření likvoru ani MR nepřispělo k pozitivní diagnóze, ale je vhodné především v diferenciální diagnóze. Průběh byl příznivý u všech nemocných.
Diskuze
MFS je vzácné onemocnění, jehož incidence není přesně známa a může se lišit v závislosti na geografii. Všeobecně se incidence odhaduje na jeden případ na jeden milion obyvatel za rok [2].
Většina epidemiologických dat je ze souborů s GBS a MFS se vyskytuje v 1–5 % GBS v Evropě a USA, ale až v 18 % na Taiwanu [3] a 25 % v Japonsku [4]. Jde ovšem o starší soubory, kdy ne všichni nemocní měli vyšetřeny antigangliosidové protilátky, takže definitivní diagnóza MFS nemusí být vždy jednoznačná. Muži bývají postiženi asi 2krát častěji než ženy. Mohou se vyskytnout ve všech věkových kategoriích včetně dětí.
Patogeneze
U MFS se jedná o autoimunitní reakci proti periferním nervům s uplatněním buněčné i protilátkové imunologické abnormality. Specifickou úlohu zde sehrávají antiglykolipidové protilátky, které postihují periferní nervový systém, zejména okohybné nervy. Buněčná imunita se u MFS jako axonální neuropatie uplatňuje méně. Stejně jako u GBS, tak i u MFS jsou imunitní mechanizmy často spouštěny předchozí infekcí. Časový interval vzniku potíží je nejčastěji 10–14 dní po infekci. Sérologicky bývá nejvíce prokázán Campylobacter jejuni a Hemophillus influenzae. Nejčastěji jsou citovány respirační infekce, méně gastrointestinální [2,4,5]. Ostatní patogeny (Staphylococcus aureus, Mycoplasma pneumoniae, Coxiella burnetii, Cytomegalovirus, Epstein-Baarové virus, Varicella zoster virus, virus spalniček) jsou také uváděny jako předcházející infekce, ale statisticky významná souvislost prokázána nebyla [5].
Molekulární mimikry se považují za hlavní mechanizmus, jak infekční agens může spustit imunitní odpověď. Jde o strukturální podobnost mezi vlastními humánními proteiny a proteiny mikroorganizmů. Specifický peptidový, karbohydrátový nebo lipidový epitop infekčního virového či bakteriálního agens má stejné sekvence jako antigenní epitop myelinu a lipopolysacharidy těchto organizmů sdílí epitopy podobné gangliosidům periferních nervů. [6–9]. Nejvíce byl studován Campylobacter jejuni. Autoimunitní ataka je důsledkem strukturální podobnosti mezi určitými molekulami lipooligo- nebo lipopolysacharidů Campylobacter jejuni a gangliosidy nervové tkáně a specifické bakteriální geny jsou stěžejní pro indukci antigangliosidových protilátek. Gen cst-II kóduje sialyltransferázový protein, který je základní pro biosyntézu gangliosidového epitopu i dalších glukosyltransferázových genů. V oblasti cst-II však existuje značný polymorfizmus. Cst-II (Asn51) je nosičem GQ1b epitopu a vyvolává MFS a produkci anti-GQ1b IgG protilátek, zatímco cst-II (Thr51) obsahuje GM1/GD1a epitopy, produkuje protilátky anti-GM1 a GD1a a GBS (obr. 1) [10–14].
![Schéma rozvoje GBS nebo MFS podle typu protilátek (upraveno podle [5]).
Při infekci Campylobacterem jejuni a produkci protilátek anti-GM1, GM1b, GD1a nebo GalNAc-GD1a dochází k postižení periferních motorických nervů, které obsahují obdobné epitopy a rozvíjí se axonální forma GBS (AMAN). Při produkci anti-GQ1b protilátek se rozvíjí MFS, protože GQ1b epitopy jsou hustě lokalizovány jen na okohybných nervech.](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image/ee3fa703d2d9325bc4a8cc919fe9aa0a.jpeg)
Protilátky proti gangliosidům
Gangliosidy patří mezi glykosfingolipidy, jejichž molekula se skládá z hydrofobního ceramidu a hydrofilního oligosacharidového řetězce obsahujícího jeden až čtyři monosacharidy. V těle je popsáno více než 100 gangliosidů, mimo mozek je nejčastější GM3, v nervových buňkách centrálního a periferního nervového systému GM1, GD1a, GD1b a GT1. V kraniálních okohybných nervech a v ganglion ciliare převažuje GQ1, který se v ostatních periferních nervech nevyskytuje. U MFS jsou podle recentních publikací [5–7,11] přítomny pozitivní protilátky IgG anti-GQ1b v séru ve více než v 90 %, v menším procentu mohou být i GD1b, GD1a, GD2 a GD3. Předpokládá se, že oftalmoplegie je důsledkem poruchy vedení vazbou anti-GQ1b protilátky na paranodální myelin kraniálních okohybných nervů [5–7,9,11,12,15]. Aktivita anti-GQ1b protilátek na nervosvalovém spojení je zřejmě způsobena přímou lézí presynaptické membrány cestou aktivace klasické komplementové cesty. Byly publikovány práce, které opakovaně prokázaly že anti-GQ1b protilátky způsobují funkční i strukturální změny na nervosvalové ploténce v experimentech na preparátech myší bránice nebo kultivovaných myších myotubách [16,17]. Otázkou je ovšem klinická relevance těchto experimentů. Nedávná studie [18] s použitím stimulační single fiber EMG prokázala, že anti-GQ1b protilátky neovlivňují nervosvalový přenos na končetinových svalech u pacientů s MFS nebo Bickerstaffovou kmenovou encefalitidou, kteří mají pozitivní anti-GQ1b protilátky a nepřítomné jiné antigangliosidové protilátky. Zpochybňuje tak některé předchozí práce [19–21], které zjistily poruchu nervosvalového přenosu u pacientů s MFS právě z toho důvodu, že nešlo o nemocné s pozitivitou pouze anti-GQ1b protilátek. Zjištěná porucha mohla být způsobena současnou přítomností protilátek anti-GM1, GM1b, GD1a, jež jsou asociovány s pouze axonální formou GBS.
Mechanizmus vlastní ataxie není zcela jasný. Selektivní postižení primárních aferentních vláken Ia je nejspíše zodpovědné za senzitivní ataxii, která se ovšem velmi podobá cerebelárnímu typu a uvažuje se o postižení spinocerebelárních drah. Na některých velkých neuronech v gangliích zadních míšních kořenů byla prokázána vazba protilátek anti-GQ1b, ale není jednoznačně potvrzeno, zda právě tyto neurony jsou spojeny se spinocerebelárními drahami. Poruchy vedení spinocerebelárními drahami podporují i některé elektrofyziologické práce [12,13]. Uvažuje se i o disproporci mezi postižením vláken Ia a naopak nejspíše nepostiženými vlákny Ib a II [2,10], ale i o funkčním postižení paralelních vláken mozečku [22]. V současné době převládá názor, že ataxie u MFS může mít jak periferní, tak i centrální (přímo mozečkovou) příčinu. S ataktickou formou GBS jsou nejčastěji spojovány protilátky anti-GD1b [12].
Odaka et al v roce 2001 [23] použili termín „anti-GQ1b protilátkový syndrom“, aby rozlišili různá onemocnění, která se mohou při detekci těchto protilátek vyskytovat: MFS, Bickerstaffova kmenová encefalitida, akutní oftalmoplegie bez ataxie, smíšená forma MFS a GBS, GBS s oftalmoplegií i klasický GBS.
U Bickerstaffovy kmenové encefalitidy se společně s oftalmoplegií a ataxií, které jsou přítomny vždy, vyskytují poruchy vědomí různého stupně, hyperreflexie nebo pyramidové spastické jevy. Předcházející infekce byla prokázána v 92 %, hyperproteinorhachie v 59 % a přítomnost anti-GQ1b protilátek v 66 %. MR prokázala hyperintenzity v zadní jámě lební, bílé hmotě nebo thalamu u 30 % pacientů, 73 % mělo neložiskovou theta-delta abnormitu na EEG. U 44 % byla abnormní i motorická neurografie (EMG) převážně s projevy axonální degenerace (38 %), v 6 % byly i projevy demyelinizace. Ke kompletní remisi došlo u 66 % pacientů během šesti měsíců. Mohou být přítomny i protilátky proti napěťově řízeným draslíkovým kanálům [24]. Na základě zhodnocení přítomnosti protilátek, předcházejících infektů, MR a neurofyziologických nálezů velkého souboru nemocných z Japonska [25] předpokládají autoři možnost kontinua a překrývání MFS s Bickerstaffovou kmenovou encefalitidou s variabilním postižení centrálního nebo periferního nervového systému.
U akutní oftalmoplegie bez ataxie jde o různou kombinaci zevní a vnitřní oftalmoplegie, která může být i jednostranná [26]. Nejčastější je oboustranná porucha abdukce, dále postižení n. III a vnitřní oftalmoplegie [27].
Klinická symptomatika MFS
Typický obraz se základní trias (oftalmoplegie, ataxie a areflexie) se obvykle rozvine během 5 až 10 dní po infektu, častěji horních cest dýchacích (76 %), méně častý bývá průjem (25 %) [25]. Z iniciálních příznaků MFS jsou nejčastější diplopie (65–78 %), poruchy chůze – trupová ataxie (32–46 %), případně i oba příznaky ve stejném dni (34 %), dysestezie v končetinách (14 %), blefaroptóza (2–4 %) nebo dysfagie (2 %) [2,4,28]. Diplopie se postupně horší a může vyústit do kompletní nebo jen zevní oftalmoplegie. Jsou popisovány i případy s pouze vnitřní oftalmoplegií, která izolovaně může MFS předcházet. Mydriáza se vyskytuje asi u 35–42 % pacientů [29,30]. Jednostranná nebo oboustranná ptóza se v průběhu vyvine asi ve 37 %. V úvodu MFS mohou být ŠOR jen snížené, ale postupně vznikne symetrická areflexie nebo hyporeflexie, která může přetrvávat po dlouhé období. Kromě okohybných nervů mohou být postiženy i další hlavové nervy, nejčastěji n. facialis (22 %) a kaudální bulbární nervy (17 %). Může být i mírná slabost končetin (svalová síla ≥ 4) – až ve 25 % [25]. Bývají pozitivní senzitivní příznaky (parestezie, dysestezie) všech končetin, ale senzitivní deficity nebývají výrazné. Jen vzácně MFS progreduje do obrazu smíšené formy s GBS a tyto případy jsou dnes klasifikovány jako GBS s oftalmoplegií nebo ataktická forma GBS [7]. Průměrná doba od prvních příznaků do jejich maximálního rozvoje je kolem 6 dnů (2–21), doba do začátku zlepšování průměrně kolem 14 dnů (3–46) a doba do úpravy symptomatiky velmi variabilní, průměrně 60 dnů (8–271) [2,4].
Elektrodiagnostika
Pro diagnózu MFS má velký význam EMG neurografie a redukce amplitud nebo nevýbavnost SNAP [31,32], ale v řadě popsaných případů byly i kondukční studie normální [25]. Vzhledem k možnosti určitého překrývání s GBS se mohou najít i lehké projevy demyelinizační nebo axonální polyneuropatie. Abnormity H reflexu, které se někdy uvádějí jako diagnosticky významné [33], považujeme za nespecifické – vesměs korelují se sníženým nebo vyhaslým reflexem šlachy Achillovy.
Vyšetření mozkomíšního moku má u MFS význam především v diferenciální diagnostice. Proteinocytologická disociace je mnohem méně výrazná nežli u GBS a u některých nemocných je nález i normální. Diagnosticky nejvýznamnější jsou zvýšené titry protilátek anti-GQ1b, které jsou přítomny v séru v akutní fázi, ale rychle klesají v korelaci s klinickou úpravou, takže někdy již za měsíc od začátku nemusí být prokazatelné [34]. Proteinocytologická disociace v likvoru byla zjištěna v souboru 123 pacientů s MFS v prvních třech týdnech v 59 %, kdežto protilátky anti-GQ1b, v 85 % [35].
MR má rovněž hlavní význam v diferenciální diagnostice. Jen vzácně se mohou zjistit hyperintenzity v T2 váženém obraze v diencefalu nebo mozečku [2,25]. U těžkých forem se může prokázat postkontrastní enhancement hlavových nervů [36].
Terapie
Názory na léčbu MFS nejsou jednoznačné. Vzhledem ke vzácnosti onemocnění nebyla dosud provedena žádná prospektivní randomizovaná studie a při léčbě se vychází ze skutečnosti, že jde o variantu GBS, a léčba je proto obdobná [37,38]. MFS je však obecně považován za „self limited“ onemocnění s většinou benigním průběhem a spontánní úpravou [2,4,7]. Mori et al [39] retrospektivně zhodnotili léčbu velké skupiny 92 nemocných s MFS, 28 bylo léčeno IVIG, 23 plazmaferézou a 41 pacientů s jen symptomatickou terapií sloužilo jako kontrolní skupina. Ve skupině léčených IVIG došlo k mírnému urychlení ústupu oftalmoplegie i ataxie (obr. 2), ale k úpravě došlo u všech nemocných. V současné době neexistují jednoznačná doporučení pro léčbu MFS [40,41]. Všechny naše nemocné jsme léčili IVIG, tedy obdobně jako léčíme všechny nemocné s těžší formou GBS. Obecně je možno léčbu IVIG doporučit hlavně u těžších forem MFS nebo určitého překryvu s GBS.
![Kaplanova-Meierova křivka zobrazuje procento pacientů s MFS léčených IVIG, plazmaferézou (PF) a kontrolní skupinu s jen symptomatickou léčbou [37]. A – ústup oftalmoplegie, B – ústup ataxie. IVIG mírně urychlil ústup oftalmoplegie (p = 0,04) i ataxie (p = 0,027).](https://www.csnn.eu/media/cache/resolve/media_object_image_large/media/image/a9534296696d7eb17d3c27932e7addaa.jpeg)
prof. MUDr. Zdeněk Ambler, DrSc.
Neurologická klinika LF UK a FN Plzeň
Alej Svobody 80
304 60 Plzeň
e-mail: ambler@fnplzen.cz
Přijato k recenzi: 26. 5. 2011
Přijato do tisku: 15. 6. 2011
Zdroje
1. Fisher M. An unusual variant of acute idiopathic polyneuritis (syndrome of ophthalmoplegia, ataxia and areflexia). N Engl J Med 1956; 255(2): 57–65.
2. Lo YL. Clinical and immunological spectrum of the Miller Fisher syndrome. Muscle Nerve 2007; 36(5): 615–627.
3. Lyu RK, Tang LM, Cheng SY, Hsu WC, Chen ST. Guillain-Barré syndrome in Taiwan: a clinical study of 167 patients. J Neurol Neurosurg Psychiatry 1997; 63(4): 494–500.
4. Mori M, Kuwabara S, Fukutake T, Yuki N, Hattori T. Clinical features and prognosis of Miller Fisher. Neurology 2001; 56(8): 1104–1106.
5. Yuki N. Infectious origins of, and molecular mimicry in, Guillain-Barré and Fisher syndromes. Lancet Infect Dis 2001; 1(1): 29–37.
6. Willison HJ, Yuki N. Peripheral neuropathies and anti-glycolipid antibodies. Brain 2002; 125(12): 2591–2625.
7. Overell JR, Willison HJ. Recent developments in Miller Fisher syndrome and related disorders. Cur Opin Neurol 2005; 18)5): 562–566.
8. Ang CW, Jacobs BC, Laman JD. The Guillain-Barré syndrome: a true case of molecular mimicry. Trends Immunol 2004; 25(2): 61–66.
9. Willison HJ. Ganglioside complexes as targets for antibodies in Miller Fisher syndrome. J Neurol Neurosurg Psychiatry 2006; 77(9): 1002–1003.
10. Yuki N. Ganglioside mimicry and peripheral nerve disease. Muscle Nerve 2007; 35(6): 691–711.
11. Kanzaki M, Kaida K, Ueda M, Morita D, Hirakawa M, Motoyoshi K et al. Ganglioside complexes containing GQ1b as targets in Miller Fisher and Guillain--Barré syndromes. J Neurol Neurosurg Psychiatry 2008; 79(10): 1148–1152.
12. Kaida K, Ariga T, Yu RK. Antiganglioside antibodies and their pathophysiological effects on Guillain--Barré syndrome and related disorders – a review. Glycobiology 2009; 19(7): 676–692.
13. Kaida K, Kusunoki S. Antibodies to gangliosides and ganglioside complexes in Guillain-Barré syndrome and Fisher syndrome: mini-review. J Neuroimmunol 2010; 223(1–2): 5–12.
14. Kivity S, Agmon-Levin N, Blank M, Shoenfeld Y. Infections and autoimmunity – friends or foes? Trends Immunol 2009; 30(8): 409–414.
15. Yuki N. Campylobacter sialyltransferase gene polymorphism directs clinical features of Guillain--Barré syndrome. J Neurochem 2007; 103 (Suppl 1): 150–158.
16. Jacobs BC, Bullens RW, O’Hanlon GM, Ang CW, Willison HJ, Plomp JJ. Detection and prevalence of α-latrotoxin-like effects of serum from patients with Guillain-Barré syndrome. Muscle Nerve 2002; 25(4): 549–558.
17. Buchwald B, Bufler J, Carpo M, Heidenreich F, Pitz R, Dudel J et al. Combined pre- and postsynaptic action of IgG antibodies in Miller Fisher syndrome. Neurology 2001; 56(1): 67–74.
18. Kuwabara S, Misawa S, Takahashi H, Sawai S, Kanai K, Nakata M et al. Anti-GQ1b antibody does not affect neuromuscular transmission in human limb muscle. J Neuroimmunol 2007; 189(1–2): 158–162.
19. Lo YL, Leoh TH, Dan YF, Lim LL, Seah A, Fook--Chong S et al. Presynaptic neuromuscular transmission defect in the Miller Fisher syndrome. Neurology 2006; 66(1): 148–149.
20. Lange DJ, Deangelis T, Sivak MA. Single-fiber electromyography shows terminal axon dysfunction in Miller Fisher syndrome: a case report. Muscle Nerve 2006; 34(2): 232–234.
21. Tomcík J, Dufek M, Hromada J, Rektor I, Bares M. Recurrent Miller Fisher syndrome with abnormal terminal axon dysfunction: a case report. Acta Neurol Belg 2007; 107(4): 112–114.
22. Lo YL, Fook-Chong S, Chan LL, Ong WY, Ratnagopal P. Electrophysiological evidence of cerebellar fiber system involvement in the Miller Fisher syndrome. J Neurol Sci 2010; 288(1–2): 49–53.
23. Odaka M, Yuki N, Hirata K. Anti-GQ1b IgG antibody syndrome: clinical and immunological range. Neurol Neurosurg Psychiatry 2001; 70(1): 50–55.
24. Tüzün E, Kürtüncü M, Lang B, İçöz S, Akman-Demir G, Eraksoy M et al. Bickerstaff’s encephalitis and Miller Fisher syndrome associated with voltage-gated potassium channel and novel anti-neuronal antibodies. Eur J Neurol 2010; 17(10): 1304–1307.
25. Ito M, Kuwabara S, Odaka M, Misawa S, Koga M, Hirata K et al. Bickerstaff’s brainstem encephalitis and Fisher syndrome form a continuous spectrum. Clinical analysis of 581 cases. J Neurol 2008; 255(5): 674–682.
26. Lee SH, Lim GH, Kim JS, Oh SY, Kim JK, Cha JK et al. Acute ophthalmoplegia (without ataxia) associated with anti-GQ1b antibody. Neurology 2008; 71(6): 426–429.
27. Fleury V, Aqallal A, Lagrange E, Besson G, Caudie C. Acute bilateral mydriasis associated with anti-GQ1b antibody. J Clin Neurosci 2010; 17(4): 514–515.
28. Špalek P, Martinka I, Jurčaga F, Richter D, Hanáčková E. Miller Fisherov syndróm – tri kazuistiky, diagnostika a liečba. Neurológia 2009; 4: 101–105.
29. Papanikolaou T, Gray C, Boothman B, Naylor G, Mariatos G. Acute bilateral ophthalmoparesis with pupilary areflexical mydriasis in Miller-Fisher syndrome treated with intravenous immunoglobulin. J Ophthalmol 2010; 2010: 291840.
30. Bae JS, Kim JK, Kim SH, Kim OK. Bilateral internal ophthalmoplegia as an initial sole manifestation of Miller Fisher syndrome. J Clin Neurosci 2009; 16(7): 963–964.
31. Fross RD, Daube JR. Neuropathy in the Miller Fisher syndrome: Clinical and electrophysiologic findings. Neurology 1987; 37(9): 1493–1498.
32. Hughes RA, Cornblath DR. Guillain-Barré syndrome. Lancet 2005; 366(9497): 1653–1666.
33. Dachy B, Deltenre P, Deconinck N, Dan B. The H reflex as a diagnostic tool for Miller Fisher syndrome in pediatric patients. J Clin Neurosci 2010; 17(3): 410–411.
34. Odaka M, Koga M, Yuki N, Susuki K, Hirata K. Longitudinal changes of anti-ganglioside antibodies before and after Guillain-Barré syndrome onset subsequent to Campylobacter jejuni enteritis. J Neurol Sci 2003; 210(1–2): 99–103.
35. Nishimoto Y, Odaka M, Hirata K, Yuki N. Usefulness of anti-GQ1b IgG antibody testing in Fisher syndrome compared with cerebrospinal fluid examination. J Neuroimmunol 2004; 148(1–2): 200–205.
36. Kiphuth IC, Saake M, Lunkenheimer J, Dörfler A, Schwab S, Kollmar R. Bilateral enhancement of the cranial nerves III–XII in severe Miller-Fisher syndrome. Eur Neurol 2009; 62(4): 252–253.
37. Lehmann HC, Hartung HP. Complementing the therapeutic armamentarium for Miller Fisher syndrome and related immune neuropathies. Brain 2008; 131(5): 1168–1170.
38. Mori M, Kuwabara S, Fukutake T, Hattori T. Plasmapheresis and Miller Fisher syndrome: analysis of 50 consecutive cases. J Neurol Neurosurg Psychiatry 2002; 72(5): 680.
39. Mori M, Kuwabara S, Fukutake T, Hattori T. Intravenous immunoglobulin therapy for Miller Fisher syndrome. Neurology 2007; 68(14); 1144–1146.
40. Overell JR, Hsieh ST, Odaka M, Yuki N, Willison HJ. Treatment for Fisher syndrome, Bickerstaff‘s brainstem encephalitis and related disorders. Cochrane Database Syst Rev 2007; 24(1): CD004761.
41. Bednařík J, Voháňka S, Ehler E, Ambler Z, Piťha J, Vencovský J et al. Standard pro léčbu pacientů s autoimunitními nervosvalovými onemocněními intravenózním lidským imunoglobulinem a plazmaferézou. Cesk Slov Neurol N 2010; 73/106(6): 579–589.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2011 Číslo 6
Nejčtenější v tomto čísle
- Miller Fisherův syndrom – čtyři vlastní pozorování a přehled současných poznatků
- Současný pohled na patofyziologii migrény
- Operační léčba poranění plexus brachialis
- Novelizace české verze Addenbrookského kognitivního testu (ACE-CZ)