
Prionový protein, jeho úloha v buněčné proliferaci, diferenciaci a vývoji nervové soustavy
Prion Protein, its Role in Cellular Proliferation, Differentiation and Nervous System Development
The cellular prion protein (PrPC) is well-known for its ability to converse into its pathological isoform, PrPTSE. Accumulation of PrPTSE in the brain is associated with pathogenesis of prion diseases. Numerous studies have suggested that PrPC has a number of physiological functions and participates in many cellular processes. However, convincing evidence is still missing. Possible functions of PrPC include a role in regulation of apoptosis, protection against oxidative stress, cell adhesion or processes of learning and memory. This protein also seems to influence cell proliferation and differentiation. The level of PrPC expression during embryonic development affects transcription of genes encoding factors involved in the regulation of stem cells pluripotency at early stages of differentiation. In the nervous system, PrPC plays an important role in neuronal development, maturation and neural circuit formation. Finally, PrPC can probably also participate in the differentiation and proliferation of tissue-specific stem cells such as neuronal, hematopoietic or myogenic precursors.
Key words:
prions – prion protein – PrPC – cell differentiation – cell proliferation – embryonic stem cells – neurons – neurogenesis
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Autoři:
Z. Hanusová 1; J. Kučerová 1,2; M. Filipová 1; Z. Jindrová 1,3; K. Holada 1
Působiště autorů:
Ústav imunologie a mikrobiologie, 1. LF UK v Praze
1; Přírodovědecká fakulta, UK v Praze
2; 3. lékařská fakulta UK v Praze
3
Vyšlo v časopise:
Cesk Slov Neurol N 2015; 78/111(4): 406-412
Kategorie:
Přehledný referát
Souhrn
Buněčný prionový protein (PrPC) je známý především jako prekurzor patologické konformační izoformy prionového proteinu PrPTSE, jejíž hromadění v mozku je spojeno s patogenezí prionových onemocnění. Výsledky četných studií naznačují, že PrPC má také řadu fyziologických funkcí a účastní se mnoha buněčných procesů; přesvědčivé důkazy nicméně stále chybí. Popsána byla možná úloha PrPC v regulaci apoptózy, ochraně proti oxidačnímu stresu, buněčné adhezi nebo procesech učení a paměti. PrPC zřejmě významně ovlivňuje také diferenciaci a proliferační aktivitu buněk. Během embryonálního vývoje má hladina exprese PrPC vliv na transkripci genů, které se podílejí na regulaci pluripotence kmenových buněk v raných fázích diferenciace. Důležitou roli hraje PrPC v nervové soustavě, kde se uplatňuje ve vývoji a zrání neuronů a při formování neuronálních obvodů. V neposlední řadě se PrPC zřejmě může podílet i na diferenciaci a proliferaci tkáňově specifických kmenových buněk, jako jsou neuronální, myogenní nebo hematopoietické prekurzory.
Klíčová slova:
priony – prionový protein – PrPC – buněčná diferenciace – buněčná proliferace – embryonální kmenové buňky – neurony – neurogeneze
Úvod – priony a prionová onemocnění
Buněčný prionový protein (PrPC) je glykoprotein lokalizovaný na povrchu téměř všech buněk v organizmu, vysoce konzervovaný mezi obratlovci [1]. Přeměna normálního prionového proteinu PrPC s převážně α‑ helikální strukturou na proteolyticky rezistentní izoformu (PrPTSE) bohatou na β‑listy je těsně spojena s patogenezí prionových onemocnění [2]. Prionová infekce se zásadně liší od onemocnění způsobených viry, viroidy či bakteriemi, pro které je esenciálním zdrojem dědičných informací nukleová kyselina. V případě prionových chorob kódování patogenního PrP nukleovou kyselinou chybí a infekční agens je tvořeno patologickým PrPTSE, který má stejnou primární strukturu jako normální PrPC a liší se od něj pouze tvarem molekuly [3]. S tím se pojí neschopnost imunitního systému rozeznat priony jako cizí, a při onemocnění tak nedochází k rozvinutí systémové imunitní odpovědi. Orgány imunitního systému naopak slouží jako místo prvotního pomnožení prionů po jejich vstupu do organizmu.
Prionové choroby, nazývané také transmisivní spongiformní encefalopatie (TSE), jsou fatální neurodegenerativní onemocnění postihující řadu savců včetně člověka. Pro tato onemocnění, jejichž hlavními klinickými příznaky jsou poruchy kognitivních a pohybových funkcí, není známa žádná účinná léčba [4]. Mezi příklady TSE patří např. bovinní spongiformní encefalopatie skotu (BSE), klusavka („scrapie“) ovcí nebo lidská onemocnění kuru a Creutzfeldt‑ Jakobova choroba (Creutzfeldt‑ Jakob Disease; CJD) [5,6]. Dosud bylo zaznamenáno okolo 220 případů tzv. variantní CJD (vCJD), jejíž vznik je spojován s přenosem BSE na člověka prostřednictvím konzumace potravin kontaminovaných priony pocházející z nakaženého skotu [7]. Lidská prionová onemocnění však nejčastěji vznikají sporadicky, tj. bez známé příčiny (~85 %); část případů (~15 %) tvoří onemocnění genetická, podmíněná mutacemi genu pro prionový protein (familiární/ genetická CJD, Gerstmann‑Sträussler‑ Scheinkerův syndrom nebo fatální familiární insomnie) [8,9]. Všechny lidské prionové choroby včetně genetických forem byly experimentálně přeneseny na laboratorní zvířata, u CJD byl navíc prokázán iatrogenní přenos prostřednictvím kontaminovaných chirurgických nástrojů a biologického materiálu [10,11]. Prionová onemocnění se vyskytují s incidencí 1– 2 případy na milion obyvatel ročně, nicméně průzkumy ukazují, že řada případů může dnešní diagnostice unikat, a existuje tak riziko podhodnocení počtu postižených jedinců [8,12]. V České republice byl mezi lety 2002– 2012 zaznamenán výskyt 138 případů lidských prionových chorob [13].
Prionová onemocnění se vyznačují dlouhou inkubační dobou. Po nástupu příznaků dochází k rychlé progresi choroby vedoucí ke smrti postiženého jedince v rozpětí několika měsíců až dvou let [14]. Mezi hlavní příznaky patří rychlý rozvoj demence, změny chování, poruchy motoriky, ataxie, myoklonus, postižení zraku a řeči [14]. Periferní infekce TSE vede k primární akumulaci PrPTSE v lymfatické tkáni, kde se protein hromadí především ve folikulárních dendritických buňkách, případně v makrofázích [15]. Přenos prionů do centrální nervové soustavy (CNS) poté probíhá parasympatickými a sympatickými nervovými vlákny [16].
Typickými neuropatologickými znaky prionových onemocnění jsou hromadění PrPTSE v mozkové tkáni ve formě amyloidu, astroglióza, degenerace neuronů a spongiformní změny v mozku [17]. Svým charakterem jsou prionové choroby podobné dalším neurodegenerativním onemocněním, jako je Alzheimerova nebo Parkinsonova nemoc, které se v populaci vyskytují daleko častěji a pro něž je také charakteristická akumulace konformačně pozměněných proteinů ve formě amyloidových depozit v mozku postižených jedinců. Při Parkinsonově chorobě dochází k akumulaci α‑ synukleinu v Lewyho tělíscích, u Azheimerovy choroby jsou plaky tvořeny proteinem amyloidem β (Aβ) [18,19]. Zásadní rozdíl mezi těmito onemocněními a prionovými chorobami je jejich přenositelnost; zatímco PrPTSE vytváří infekční partikule schopné infikovat jiné jedince a možný je i přenos mezidruhově, epidemiologické studie zatím nenaznačují, že by se např. Alzheimerova choroba mohla v lidské populaci šířit infekcí [20]. Bylo však ukázáno, že experimentální přenos Alzheimerovy nemoci na geneticky modifikovaná laboratorní zvířata prostřednictvím mozkového homogenátu pacienta možný je a infekční částice se v tomto případě skládá z proteinu Aβ [18]. Klasifikace neurodegenerativních proteinopatií, často také označovaných jako tzv. konformační choroby, je v současnosti široce diskutována. Přítomnost patologicky sbalených proteinů schopných šířit se v rámci organizmu, společná jak prionovým, tak dalším neurodegenerativním proteinopatiím, je na jedné straně vnímána jako definující vlastnost pro tuto skupinu onemocnění; Alzheimerova nebo Parkinsonova nemoc by tedy také mohly být považovány za „prionové“ [21]. Na straně druhé nedostatek dat podporujících hypotézu, že by i další proteiny mohly tak jako PrP hrát za přirozených podmínek roli infekčních agens, vede k nutnosti rozlišení mezi dalšími neurodegenerativními proteinopatiemi a prionovými chorobami jako takovými [22]. Hlubší porozumění této problematice je nutné nejen pro objasnění vztahu mezi oběma skupinami chorob, ale také pro nalezení jejich efektivní terapie.
Studie zaměřené na odhalení fyziologické úlohy PrPC naznačily, že protein se v organizmu účastní řady procesů; ucelená představa o jeho roli je však stále nejasná [23]. Cílem tohoto přehledného článku je poskytnout stručné shrnutí informací o možných fyziologických úlohách PrPC se zvláštním zaměřením na jeho roli v procesech buněčné diferenciace a proliferace.
Prionový protein a jeho struktura
PrPC, buněčná varianta prionového proteinu, je glykoprotein o velikosti 30– 35 kDa asociovaný s membránou buněk prostřednictvím glykosylfosfatidylinositolové (GPI) kotvy. U lidí je PrPC kódován genem PRNP, který se nachází na krátkém raménku 20. chromozomu (20q13). PRNP kóduje prekurzorový protein o velikosti 253 aminokyselinových (AK) zbytků. Při posttranslačních modifikacích PrPC dochází k odštěpení 22 AK z N‑ konce proteinu a po připojení GPI kotvy v endoplazmatickém retikulu také prvních 23 C‑ koncových AK; maturovaný protein má tedy 208 AK [24].
Sekundární strukturu PrPC tvoří několik domén (obr. 1). Neuspořádaná N‑terminální doména obsahuje sérii pěti oktarepetic bohatých na histidin, jež mohou sloužit jako vazebné místo pro dvojmocné kationty (Cu2+, Zn2+, Ni2+, Mn2+) [25]. Centrální část prionového proteinu je tvořena oblastí s nabitými aminokyselinami následovanou hydrofóbní, zdánlivě transmembránovou oblastí, která za normálních podmínek není využita [26]. C‑terminální doména PrPC je organizována do tří α‑ šroubovic (aminokyselinové zbytky 144– 154, 173– 194 a 200– 228 u lidského PrPC) a jednoho antiparalelního β‑ skládaného listu (β‑ řetězce tvořené AK zbytky 128– 131 a 161– 164) [27]; mezi aminokyselinami Cys179 a Cys214 se nachází disulfidový můstek [28]. Tato doména obsahuje dvě N‑ glykosylační místa (Asp181 a Asp197); PrPC se tak v buňce může nacházet v různých formách – neglykosylovaný, mono‑ či diglykosylovaný [24].
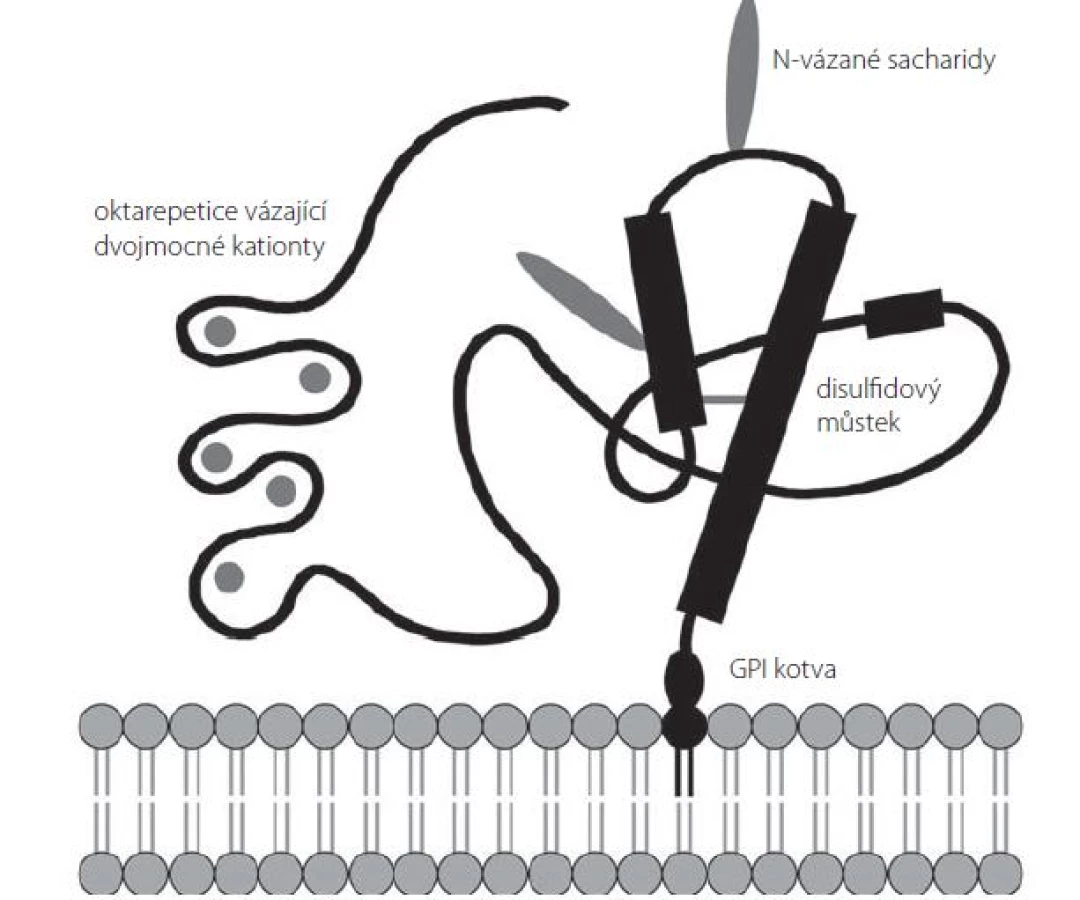
Dosud bylo popsáno více než 30 různých bodových mutací genu PRNP spojených s výskytem genetických forem prionových onemocnění. Nejčastěji se jedná o substituční mutace vedoucí k záměně kódované aminokyseliny v C‑terminální doméně PrPC mezi AK 178 a 217. Časté jsou u těchto nemocí také případy několikanásobného zmnožení či delece oktapeptidů v sérii oktarepetic [29]. Celosvětově nejběžnější mutace je aminokyselinová záměna E200K, která je spojena s genetickou formou CJD a dosahuje neobvykle vysokého výskytu na Slovensku, kde tvoří více než 65 % všech zaznamenaných případů CJD [30].
Většina molekul PrPC je lokalizována na membráně v lipidových raftech a kontinuálně podstupuje cirkulaci mezi povrchem buňky a endozomy prostřednictvím endocytózy [31]. Kromě maturované formy proteinu se v buňce mohou nacházet i jeho zkrácené varianty N1 (AK zbytky 23– 110), C1 (AK zbytky 111– 253) a také varianta postrádající GPI kotvu, PrP(∆GPI). Fragment C1 je stejně jako PrPC vázaný na membránu GPI kotvou a výsledky studií naznačují, že se jedná o produkt metabolizmu PrPC na membráně; sekretované fragmenty N1 a PrP(DGPI) jsou pak produkty štěpení PrPC. Fyziologická varianta PrP bez GPI kotvy zřejmě může vznikat i alternativním sestřihem transkriptu PRNP genu [32]. Úloha těchto fragmentů v organizmu není doposud plně objasněna. Fragment N1 může působit neuroprotektivně, fragment C1 je považován za inhibitor prionové infekce; zároveň však bylo ukázáno, že za určitých podmínek může C1 zvyšovat citlivost buněk k apoptotickým stimulům [33].
Normální prionový protein PrPC se při prionových onemocněních mění na patologickou variantu PrPTSE. Obě izoformy mají shodnou primární sekvenci aminokyselin, rozdíl je v jejich sekundární struktuře [34]. Konformační změny N‑ konce a centrální části molekuly PrPC vedou k vytvoření struktury PrPTSE bohaté na β‑listy, což ji činí částečně odolnou vůči proteolýze a umožňuje její ukládání ve tkáních; C‑ konec zůstává většinou nezměněn [35,36]. Tato přeměna probíhá jako posttranslační děj, který se odehrává na povrchu membrán nebo v endomembránovém systému. Strukturu patologické varianty PrPTSE zřejmě ovlivňuje také poměr zastoupení jednotlivých glykoforem prionového proteinu ve vznikající infekční částici. Glykosylace je tedy jedním z faktorů, který může přispět k vysvětlení existence prionových onemocnění s různými fenotypy v rámci jednoho živočišného druhu [37]. Zatímco PrPC lze plně rozštěpit působením proteinázy K, patologická varianta proteinu je vůči štěpení odolná a dochází k odštěpení pouze N‑ koncové části proteinu [38]. Této vlastnosti PrPTSE se využívá při diagnostice prionových chorob: detekce neštěpitelného zbytku PrPTSE o velikosti 27– 30 kDa (PrPres) pomocí specifických protilátek metodami imunohistochemie či Western blotu slouží jako nástroj pro potvrzení prionové infekce [39,40]. PrPTSE je odolný vůči běžně používaným metodám sterilizace (autoklávování při teplotě 121 °C, působení UV či ionizujícího záření); pro inaktivaci infekčního materiálu je tedy třeba používat účinnějších postupů, zahrnujících např. inaktivaci prionů pomocí 1 M NaOH nebo 2% chlornanu sodného [41].
Fyziologická úloha buněčného prionového proteinu
Buněčný prionový protein, jehož exprese začíná již v rané embryogenezi, je přítomen na povrchu řady buněk v organizmu [42]. Nejhojnější je jeho zastoupení v nervové soustavě; zde se vyskytuje především v neuronech, v nižší míře pak v gliových buňkách. Jeho výskyt v CNS však není homogenní; jak její jednotlivé oblasti, tak i funkční typy buněk se v expresi PrPC liší [1]. Přítomnost prionového proteinu byla identifikována i v buňkách dalších tkání, jež zahrnují periferní nervy, lymforetikulární systém, gastrointestinální trakt, varlata, ledviny či svaly [43]. PrPC je v různé míře exprimován také v krevních buňkách a buňkách imunitního systému [44,45].
Prionový protein je lokalizován na povrchu buněk především v oblastech s vysokou koncentrací signálních molekul, tzv. lipidových raftech, což naznačuje jeho možnou úlohu jako receptoru v procesu signální transdukce [46]. Vzhledem k tomu, že PrPC nemá intracelulární doménu a nemůže tedy aktivovat signální kaskády uvnitř buňky přímo, bylo věnováno velké úsilí identifikaci jak jeho potenciálních ligandů, tak molekul, které by mohly působit jako přenašeč signálu od PrPC směrem do nitra buňky. Bylo zjištěno, že PrPC vykazuje afinitu k celé řadě buněčných proteinů; fyziologický význam vazeb však zůstává většinou neznámý [1,47]. Nedávné studie odhalily schopnost PrPC působit jako specifický receptor pro oligomery amyloidu β (Aβ), který zároveň neváže monomery ani fibrily tohoto proteinu. Prionový protein se tak zřejmě může podílet na synaptické toxicitě zprostředkované Aβ oligomery, jež přispívá k rozvoji a progresi Alzheimerovy choroby [48].
Fyziologická funkce buněčného prionového proteinu nebyla dosud dostatečně objasněna. Uvažuje se o jeho úloze v řadě dějů zahrnujících například neuroprotekci, regulaci apoptózy, ochranu před oxidačním stresem, utváření a funkci synapsí, proces učení a paměti, ovlivnění cirkadiálních rytmů, metabolizmus mědi, buněčnou proliferaci a diferenciaci a další procesy [49].
Jednou z popsaných rolí prionového proteinu je jeho funkce jako antiapoptotického regulátoru. Bylo ukázáno, že míra exprese PrPC ovlivňuje citlivost neuronů vůči apoptóze spuštěné proteinem Bax [50]. Interakce PrPC s proteinem STI1 v retinálních neuronech zřejmě také indukuje neuroprotektivní signály [51]. Několik studií naznačilo, že PrPC se podílí na ochraně buněk před oxidačním poškozením; tato funkce je často spojována se schopností prionového proteinu vázat měďnaté ionty. V souladu s tím byla kultura neuronů postrádající PrPC náchylnější vůči působení volných iontů mědi indukujících vznik reaktivních forem kyslíku [52]; PrP‑ deficientní buňky jsou citlivější vůči oxidačnímu stresu obecně a vykazují sníženou aktivitu superoxid dismutázy, která je důležitým buněčným antioxidantem [53]. Prionový protein byl spojován i s metabolizmem mědi, nicméně výsledky studií ukazují, že PrPC zřejmě nepatří mezi klíčové molekuly, jež se na regulaci tohoto děje podílejí [23]. Zásadní úlohu má zřejmě prionový protein v hipokampu, kde jeho interakce s lamininem ovlivňuje neurální plasticitu důležitou pro utváření krátkodobé paměti a prostorovou orientaci [54,55].
Pro studium fyziologické funkce proteinů jsou kromě tkáňových kultur [56] s výhodou využívány transgenní myši s vyřazeným genem, jenž danou bílkovinu kóduje. Také v případě prionového proteinu bylo připraveno několik kmenů myší, které jsou pro tento protein deficientní [57]. Nejvýznamnějším fenotypovým projevem u těchto myší je jejich rezistence k prionové infekci, což je v souladu s prionovou hypotézou – nepřítomnost molekul PrPC ve tkáních, jež by mohly sloužit jako substrát pro generaci patologického PrPTSE, chrání myši před nákazou priony [58]. S výjimkou této vlastnosti však myši nevykazují jiné závažné fenotypové změny, jako jsou vývojové a anatomické abnormality, snížená fertilita nebo délka života [59]. To naznačuje, že funkce PrPC je za normálních podmínek zřejmě kompenzována prostřednictvím exprese jiných proteinů.
Prionový protein v embryogenezi a jeho úloha v embryonálních kmenových buňkách
Expresi buněčného prionového proteinu je možné detekovat již v raných stadiích embryogeneze. Studium embryonálního vývoje myší ukázalo, že PrPC se začíná vysoce exprimovat u embryí starých 7,5– 8,5 dne, a to především v postmitotických neuronálních buňkách ventrikulární zóny, jež prošly diferenciací [60]. V dalších dnech embryonálního vývoje se exprese neurálního PrPC rozšiřuje a ode dne 13,5 lze expresi detekovat také v jiných vyvíjejících se částech embrya, jako je např. střevo, ledviny, či dentální lamina, a v maternálních buňkách placenty, amnionu a mezodermální vrstvě žloutkového obalu [42,61]. Studium časného embryonálního vývoje skotu ve dnech 27– 39 ukázalo, že stejně jako u myší i zde je vysoká exprese PrPC v diferencovaných neuronálních buňkách, nacházejících se v marginální vrstvě neuroepitelia. Naproti tomu v periventrikulární zóně, kde se nacházejí mitoticky aktivní progenitorové buňky, exprese PrPC chybí [62]. To naznačuje, že prionový protein by mohl hrát úlohu v procesu neurální diferenciace.
Tato hypotéza byla podpořena výsledky experimentů s myšími embryonálními kmenovými buňkami (mESC). Ty ukázaly, že exprese PrPC v mESC negativně koreluje s hladinou transkripčního faktoru Oct4 udržujícího pluripotenci buněk [63]. Zatímco množství Oct4 s postupnou diferenciací buněk klesá, produkce PrPC se s rostoucí mírou diferenciace zvyšuje. PrPC také ovlivňuje transkripci genu Nanog, jež se rovněž podílí na udržení pluripotentního stavu mESC v raných fázích diferenciace [64]. Úloha prionového proteinu byla popsána i v lidských embryonálních kmenových buňkách (hESC). V nediferencovaných hESC není PrPC detekován a jeho množství se zvyšuje až při spontánní diferenciaci buněk [65]. Také ektopická exprese PrPC indukuje u pluripotentních hESC diferenciaci [66].
Prionový protein se zřejmě může podílet na regulaci diferenciace nejen pluripotentních buněk, ale i dalších buněk. Snížení exprese PrPC prostřednictvím siRNA vede ke zpoždění vývoje nestin‑pozitivních neurálních progenitorů [63]. Umlčení exprese PrPC prostřednictvím siRNA, vyvolané u hESC s již probíhající spontánní diferenciací, vedlo u těchto buněk ke změnám dynamiky buněčného cyklu a také rovnováhy mezi vývojem jednotlivých zárodečných linií. Diferenciace směrem k ektodermální linii, jež dává vznik i neuroektodermu, byla v tomto případě potlačena [66]. Zajímavé výsledky přinesl experiment s využitím rekombinantního prionového proteinu (rPrP). Přidání rPrP ke kultuře hESC vedlo ke zpomalení diferenciace a buňky byly schopné udržet si svou růstovou morfologii a proliferační aktivitu [65]. Zatím není jasné, jaký je mechanizmus tohoto jevu. Bylo však ukázáno, že overexprese PrPC v již diferencujících hESC vede k inhibici diferenciace a pomáhá buňkám udržovat si vysokou proliferační aktivitu, jednu ze základních charakteristik nediferencovaných kmenových buněk [66]. Úloha prionového proteinu v embryonálním vývoji je tedy zřejmě velmi komplexní a PrPC představuje důležitou regulační molekulu, jež se účastní řady probíhajících dějů.
Funkce prionového proteinu ve vývoji nervové soustavy
Během embryonálního vývoje nervové soustavy je fyziologická forma PrPC silně exprimována především v CNS a v menším množství také v periferních tkáních [42]. Imunohistochemická detekce odhalila přítomnost PrPC v lidském předním mozku počínaje 11. týdnem embryonálního vývoje [67]. Po jeho ukončení zůstává PrPC výrazně exprimován především v neuronech, zatímco v gliových buňkách (mikroglie, astrocyty a oligodendrocyty) je jeho množství výrazně nižší [67,68]. To naznačuje, že regulace hladiny exprese PrPC by mohl být jeden z faktorů ovlivňujících specifickou diferenciaci a finální fenotyp neurálních kmenových buněk, z nichž neurony a gliové buňky vznikají [69,70]. Tato hypotéza byla podpořena studiem PrP‑ deficientních myší, jejichž neurální prekurzorové buňky dozrávaly pomaleji než v případě buněk myší exprimujících PrPC. Overexprese PrPC naopak vedla ke zvýšené proliferaci neurálních prekurzorů [68].
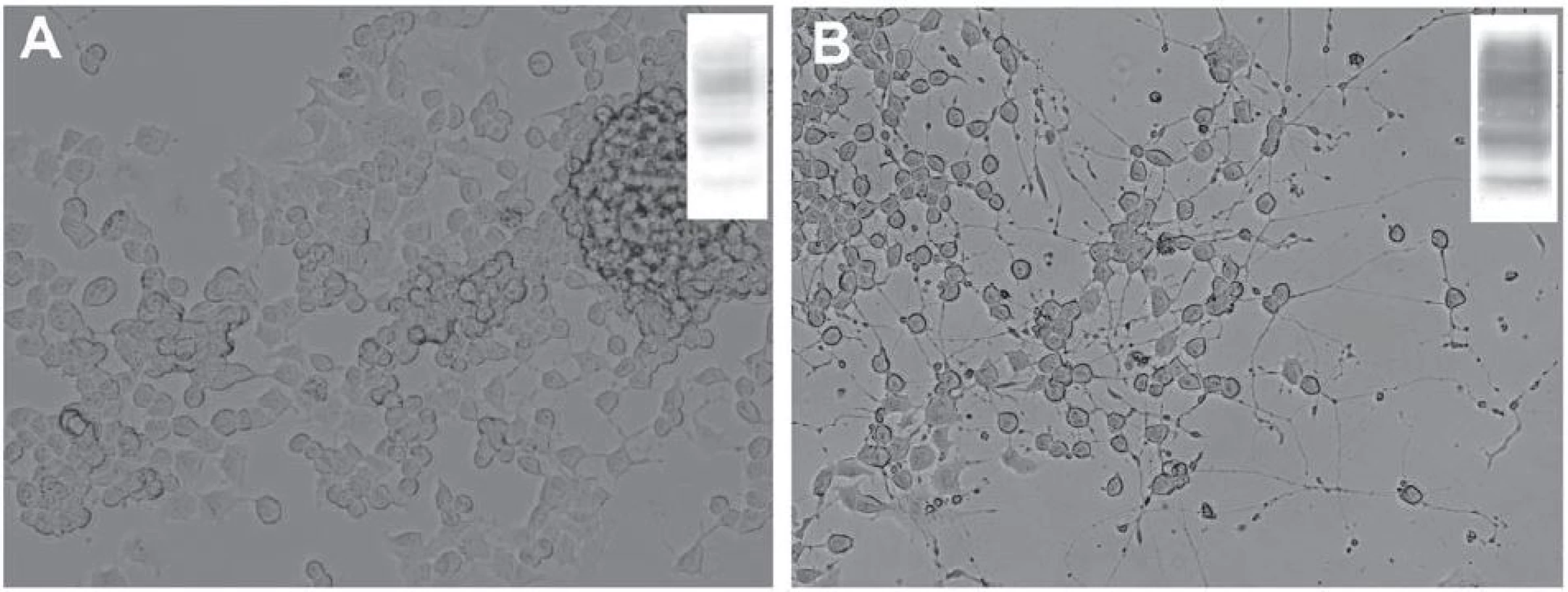
V dospělém mozku byla úloha PrPC studována v neurogenních oblastech – gyru dentatu hipokampu a subventrikulární zóně umístěné v laterálních stěnách postranních komor, kde dochází k produkci nových buněk ze stávajících prekurzorů i v dospělosti [71– 73]. Tyto nestin‑pozitivní prekurzory jsou schopné dozrávat do neuronů, astrocytů a oligodendrocytů [74]. Během diferenciace neuronů z nestin‑pozitivních prekurzorů dochází k postupnému zvyšování exprese PrPC, která je maximální u postmitotických neuronů [68]. Podobný nárůst exprese PrPC jsme zaznamenali i in vitro po indukci diferenciace myší neuronální nádorové linie (obr. 2). Hladina buněčného prionového proteinu ovlivňuje rychlost samotného zrání neuronů a uplatňuje se ve vývoji a zrání axonů, dendritů, synapsí a formování neuronálních obvodů [68,75]. Vysoká hladina PrPC na povrchu synapsí je udržována také v dospělosti a prionový protein se zřejmě podílí na celé řadě dějů, jež správnou funkci synapsí zajišťují a kontrolují [1,67]. Uvažuje se také o úloze buněčného prionového proteinu v proliferaci a diferenciaci oligodendrocytů a při maturaci astrocytů. Bylo ukázáno, že tyto děje mohou být u myší ovlivněny prostřednictvím vyřazení či naopak zvýšení exprese PrPC [76,77].
K ovlivnění neurogeneze dochází i v průběhu prionových onemocnění. Patologická izoforma prionového proteinu PrPTSE se v myších mozcích hojně vyskytuje i v neurogenních nikách. U kmenových buněk dospělého mozku PrPTSE působí narušení neuronální diferenciace a snížení počtu nezralých a dospělých neuronů ve srovnání s buňkami mozku neovlivněného PrPTSE [73]. Dochází také k narušení exprese synaptických proteinů, které jsou nezbytné pro správné fungování chemických i elektrických synapsí [78,79]. Dosavadní studie tedy ukazují, že fyziologická i patologická varianta PrP se významnou mírou podílejí na vývojových procesech probíhajících v nervové soustavě.
Vliv prionového proteinu na diferenciaci a proliferaci tkáňově specifických kmenových buněk
Buněčný prionový protein je v organizmu exprimován v celé řadě tkání; studována je tedy i jeho úloha v diferenciaci a proliferaci buněk, jež nejsou neurálního původu. Výrazné zvýšení exprese PrPC provází diferenciaci mESC směrem ke kardiomyocytům [80] a PrPC může sloužit jako specifický povrchový marker kardiomyogenních prekurzorů [81]. Mechanizmus regenerace určitých tkání po poškození vykazuje podobnost s procesem jejich morfogeneze; tak je tomu i v případě svalové tkáně. V průběhu myogeneze dochází ke změnám hladiny exprese PrPC, což ukazuje na jeho možnou úlohu v tomto ději [82]. Při studiu poškození svalové tkáně s využitím PrP‑ deficientních myší pak bylo zjištěno, že nepřítomnost PrPC je spojena s prodloužením doby hojení svalu; ovlivněna je v tomto případě proliferační a diferenciační aktivita myogenních prekurzorových buněk [83].
Expresi PrPC lze detekovat v různých typech krevních buněk a jejich prekurzorů. Zajímavá zjištění přinesly experimenty porovnávající expresi PrPC v krvi různých druhů organizmů. Prostřednictvím průtokové cytometrie bylo zjištěno, že hladina exprese buněčného prionového proteinu v bílých a červených krvinkách i krevních destičkách se výrazně liší napříč spektrem používaných modelových organizmů (myš, křeček, různé druhy opic) a ani jeden z těchto druhů nekopíruje expresní profil PrPC v lidské krvi [84,85]. To s sebou přináší nutnost obezřetnosti při interpretaci a zobecňování výsledků, získaných zejména z experimentů prováděných na laboratorních hlodavcích [86].
Vysoká množství buněčného prionového proteinu exprimují i hematopoietické kmenové buňky (Hematopoietic Stem Cells; HSC) [87]. Během diferenciace těchto buněk dochází k rozrůznění množství PrPC v jednotlivých typech prekurzorových buněk. Studium lidských krevních elementů ukázalo, že v průběhu vývoje granulocytů dochází ke snižování exprese prionového proteinu, zatímco u monocytů a lymfocytů je exprese PrPC zachována [88]. Také myší lymfoidní prekurzory exprimují buněčný prionový protein; na rozdíl od lidských buněk však maturované lymfocyty expresi PrPC ztrácejí [87]. Podobných výsledků bylo dosaženo i v případě studia myší erytropoezy. Časná fáze diferenciace myších erytroidních prekurzorů je spojena se zvýšením exprese PrPC, jehož hladina pak s postupnou maturací klesá [89]. V lidských červených krvinkách lze buněčný prionový protein detekovat i po dokončení jejich vývoje [90].
Výsledky studií zahrnujících PrP‑ deficientní myši naznačují, že absence PrPC v myších tkáních nevyvolává závažné defekty v hematopoeze. Jinak tomu však je při vystavení buněk stresovým podmínkám. HSC neexprimující buněčný prionový protein nejsou při sériových transplantacích schopné zrekonstruovat kostní dřeň PrP‑ deficientních myší po ozáření a tento stav je pro myši letální [91]. Prionový protein se tedy zřejmě nepodílí jen na diferenciačních procesech, ale u HSC hraje také důležitou roli v udržení dlouhodobé schopnosti sebeobnovy buněk.
Závěr
Buněčný prionový protein (PrPC) se kromě své nezaměnitelné úlohy v patogenezi prionových onemocnění zřejmě také podílí na modulaci řady dalších, fyziologických dějů. Poznatky posledních let poukazují na roli PrPC v procesech proliferace a diferenciace neurálních buněk. Exprese PrPC začíná již v časných fázích embryogeneze savců. Zatímco progenitorové buňky vyvíjejícího se embrya expresi PrPC postrádají, již diferencované neurální buňky vykazují vysokou hladinu PrPC, což ukazuje na spojení prionového proteinu s regulací neurální diferenciace. Množství exprimovaného PrPC se mění i při produkci nových buněk v dospělosti. Maturace nestin‑pozitivních neurálních kmenových buněk při vývoji neuronů je spojena s postupným zvyšováním exprese PrPC a zralé neurony exprimují vysoké hladiny prionového proteinu. Uvažuje se o úloze PrPC při utváření neurálních obvodů a v neurální komunikaci. U maturovaných gliových buněk je naproti tomu exprese PrPC minimální. Modulace hladiny exprese PrPC tak zřejmě představuje jeden z klíčových faktorů, které rozhodují o osudu buňky. Detailní porozumění těmto mechanizmům by mohlo přispět k pochopení patogeneze prionových chorob a vést k navržení nových terapeutických strategií pro neurodegenerativní onemocnění.
Grantová agentura České republiky – projekt č. P303/12/1791. Grantová agentura Univerzity Karlovy v Praze – projekt č. 1200213.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Přijato k recenzi: 18. 2. 2015
Přijato do tisku: 4. 6. 2015
doc. Ing. Karel Holada, Ph.D.
Ústav imunologie a mikrobiologie
1. LF UK v Praze
Studničkova 7
128 00 Praha 2
e-mail: karel.holada@lf1.cuni.cz
Zdroje
1. Linden R, Martins VR, Prado MA, Cammarota M, Izquierdo I, Brentani RR. Physiology of the prion protein. Physiol Rev 2008; 88(2): 673– 728. doi: 10.1152/ physrev.00007.2007.
2. Priola SA, Vorberg I. Molecular aspects of disease pathogenesis in the transmissible spongiform encephalopathies. Mol Biotechnol 2006; 33(1): 71– 88.
3. Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216(4542): 136– 144.
4. Marandi Y, Farahi N, Sadeghi A, Sadeghi‑ Hashjin G. Prion diseases – current theories and potential therapies: a brief review. Folia Neuropathol 2012; 50(1): 46– 49.
5. Colby DW, Prusiner SB. Prions. Cold Spring Harb Perspect Biol 2011; 3(1): a006833. doi: 10.1101/ cshperspect.a006833.
6. Gdovinová Z. Creutzfeldtova‑ Jakobova choroba. Cesk Slov Neurol N 2013; 76/ 109(2): 138– 153.
7. Will RG, Ironside JW, Zeidler M, Cousens SN, Estibeiro K, Alperovitch A et al. A new variant of Creutzfeldt‑ Jakob disease in the UK. Lancet 1996; 347(9006): 921– 925.
8. Aguzzi A, Calella AM. Prions: Protein aggregation and infectious diseases. Physiol Rev 2009; 89(4): 1105– 1152. doi: 10.1152/ physrev.00006.2009.
9. Rusina R, Fiala J, Holada K, Matejckova M, Novakova J,Ampapa R et al. Gerstmann‑Sträussler‑ Scheinker syndrome with the P102L pathogenic mutation presenting as familial Creutzfeldt‑ Jakob disease: a case report and review of the literature. Neurocase 2013; 19(1): 41– 53. doi: 10.1080/ 13554794.2011.654215.
10. Collins S, McLean CA, Masters CL. Gerstmann‑Sträussler‑ Scheinker syndrome, fatal familial insomnia, and kuru: a review of these less common human transmissible spongiform encephalopathies. J Clin Neurosci 2001; 8(5): 387– 397.
11. Brown P, Brandel JP, Sato T, Nakamura Y, MacKenzie J, Will RG et al. Iatrogenic Creutzfeldt‑ Jakob disease, final assessment. Emerg Infect Dis 2012; 18(6): 901– 907. doi: 10.3201/ eid1806.120116.
12. Head MW. Human prion diseases: molecular, cellular and population biology. Neuropathology 2013; 33(3): 221– 236. doi: 10.1111/ neup.12016.
13. Rohan Z, Parobkova E, Johanidesova S, Koukolik F, Matej R, Rusina R. Lidksé prionové nemoci v České republice – 10 let zkušeností s diagnostikou. Cesk Slov Neurol N 2013; 76/ 109(3): 300– 306.
14. Imran M, Mahmood S. An overview of human prion diseases. Virol J 2011; 8: 559. doi: 10.1186/ 1743‑ 422X‑ 8‑ 559.
15. O‘Connor T, Frei N, Sponarova J, Schwarz P, Heikenwalder M, Aguzzi A. Lymphotoxin, but not TNF, is required for prion invasion of lymph nodes. PLoS Pathog 2012; 8(8): e1002867. doi: 10.1371/ journal.ppat.1002867.
16. Mabbott NA, Macpherson GG. Prions and their lethal journey to the brain. Nat Rev Microbiol 2006; 4(3): 201– 211.
17. Soto C, Satani N. The intricate mechanisms of neurodegeneration in prion diseases. Trends Mol Med 2011; 17(1): 14– 24. doi: 10.1016/ j.molmed.2010.09.001.
18. Prusiner SB. A unifying role for prions in neurodegenerative diseases. Science 2012; 336(6088): 1511– 1513. doi: 10.1126/ science.1222951.
19. Goedert M, Clavaguera F, Tolnay M. The propagation of prion‑like protein inclusions in neurodegenerative diseases. Trends Neurosci 2010; 33(7): 317– 325. doi: 10.1016/ j.tins.2010.04.003.
20. Schmidt C, Karch A, Korth C, Zerr I. On the issue of transmissibility of Alzheimer disease a critical review. Prion 2012; 6(5): 447– 452. doi: 10.4161/ pri.22502.
21. Prusiner SB. Biology and genetics of prions causing neurodegeneration. Annu Rev Genet 2013; 47: 601– 623. doi: 10.1146/ annurev‑ genet‑ 110711‑ 155524.
22. Ashe KH, Aguzzi A. Prions, prionoids and pathogenic proteins in Alzheimer disease. Prion 2013; 7(1): 55– 59. doi: 10.4161/ pri.23061.
23. Westergard L, Christensen HM, Harris DA. The cellular prion protein (PrP(C)): its physiological function and role in disease. Biochim Biophys Acta 2007; 1772(6): 629– 644.
24. Yusa S, Oliveira‑ Martins JB, Sugita‑ Konishi Y, Kikuchi Y. Cellular prion protein: from physiology to pathology. Viruses 2012; 4(11): 3109– 3131. doi: 10.3390/ v4113109.
25. Choi CJ, Kanthasamy A, Anantharam V, Kanthasamy AG.Interaction of metals with prion protein: possible role of divalent cations in the pathogenesis of prion diseases. Neurotoxicology 2006; 27(5): 777– 787.
26. Holada K, Simak J, Risitano AM, Maciejewski J, Young NS,Vostal JG. Activated platelets of patients with paroxysmal nocturnal hemoglobinuria express cellular prion protein. Blood 2002; 100(1): 341– 343.
27. Knaus KJ, Morillas M, Swietnicki W, Malone M, Surewicz WK, Yee VC. Crystal structure of the human prion protein reveals a mechanism for oligomerization. Nat Struct Biol 2001; 8(9): 770– 774.
28. Zahn R, Liu AZ, Luhrs T, Riek R, Von Schroetter C, Garcia FL et al. NMR solution structure of the human prion protein. Proc Natl Acad Sci U S A 2000; 97(1): 145– 150.
29. Jeong BH, Kim YS. Genetic studies in human prion diseases. J Korean Med Sci 2014; 29(5): 623– 632. doi: 10.3346/ jkms.2014.29.5.623.
30. Mitrova E, Kosorinova D, Gajdos M, Sebekova K, Tomeckova I. A pilot study of a genetic CJD risk factor (E200K) in the general Slovak population. Eur J Epidemiol 2014; 29(8): 595– 597. doi: 10.1007/ s10654‑ 014‑ 9937‑ 9.
31. Taylor DR, Hooper NM. The prion protein and lipid rafts (review). Mol Membr Biol 2006; 23(1): 89– 99.
32. Kikuchi Y, Kakeya T, Nakajima O, Sakai A, Ikeda K, Yamaguchi N et al. Hypoxia induces expression of a GPI‑ anchorless splice variant of the prion protein. FEBS J 2008; 275(11): 2965– 2976. doi: 10.1111/ j.1742‑ 4658.2008.06452.x.
33. Liang JJ, Kong QZ. Alpha‑ cleavage of cellular prion protein. Prion 2012; 6(5): 453– 460. doi: 10.4161/ pri.22511.
34. Stahl N, Prusiner SB. Prions and prion proteins. FASEB J 1991; 5(13): 2799– 2807.
35. Pan KM, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D et al. Conversion of alpha‑ helices into beta‑sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci U S A 1993; 90(23): 10962– 10966.
36. Safar J, Roller PP, Gajdusek DC, Gibbs CJ. Conformational transitions, dissociation and unfolding of scrapie amyloid (prion) protein. J Biol Chem 1993; 268(27): 20276– 20284.
37. Aguzzi A, Heikenwalder M, Polymenidou M. Insights into prion strains and neurotoxicity. Nat Rev Mol Cell Biol 2007; 8(7): 552– 561.
38. Bendheim PE, Bolton DC. A 54- kDa normal cellular protein may be the precursor of the scrapie agent protease‑resistant protein. Proc Natl Acad Sci U S A 1986; 83(7): 2214– 2218.
39. Grassi J, Maillet S, Simon S, Morel N. Progress and limits of TSE diagnostic tools. Vet Res 2008; 39(4): 33. doi: 10.1051/ vetres:2008009.
40. Dvořáková E, Holada K. Konformačně specifické protilátky a diagnostika prionových chorob. Cesk Slov Neurol N 2012; 75/ 108(3): 283– 290.
41. Sakudo A, Ano Y, Onodera T, Nitta K, Shintani H, Ikuta K et al. Fundamentals of prions and their inactivation (review). Int J Mol Med 2011; 27(4): 483– 489. doi: 10.3892/ ijmm.2011.605.
42. Manson J, West JD, Thomson V, Mcbride P, Kaufman MH,Hope J. The prion protein gene: a role in mouse embryogenesis. Development 1992; 115(1): 117– 122.
43. Ford MJ, Burton LJ, Morris RJ, Hall SM. Selective expression of prion protein in peripheral tissues of the adult mouse. Neuroscience 2002; 113(1): 177– 192.
44. Vostal JG, Holada K, Simak J. Expression of cellular prion protein on blood cells: Potential functions in cell physiology and pathophysiology of transmissible spongiform encephalopathy diseases. Transfus Med Rev 2001; 15(4): 268– 281.
45. Nitta K, Sakudo A, Masuyama J, Xue GG, Sugiura K, Onodera T. Role of cellular prion proteins in the function of macrophages and dendritic cells. Protein Pept Lett 2009; 16(3): 239– 246.
46. Brouckova A, Holada K. Cellular prion protein in blood platelets associates with both lipid rafts and the cytoskeleton. Thromb Haemost 2009; 102(5): 966– 974. doi: 10.1160/ TH09‑ 02‑ 0074.
47. Didonna A. Prion protein and its role in signal transduction. Cell Mol Biol Lett 2013; 18(2): 209– 230. doi: 10.2478/ s11658‑ 013‑ 0085‑ 0.
48. Gunther EC, Strittmatter SM. Beta‑amyloid oligomers and cellular prion protein in Alzheimer‘s disease. J Mol Med (Berl) 2010; 88(4): 331– 338. doi: 10.1007/ s00109‑ 009‑ 0568‑ 7.
49. Aguzzi A, Baumann F, Bremer J. The prion‘s elusive reason for being. Annu Rev Neurosci 2008; 31: 439– 477. doi: 10.1146/ annurev.neuro.31.060407.125620.
50. Roucou X, Leblanc AC. Cellular prion protein neuroprotective function: implications in prion diseases. J Mol Med (Berl) 2005; 83(1): 3– 11.
51. Zanata SM, Lopes MH, Mercadante AF, Hajj GNM, Chiarini LB, Nomizo R et al. Stress‑ inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. EMBO J 2002; 21(13): 3307– 3316.
52. Brown DR, Schmidt B, Kretzschmar HA. Effects of copper on survival of prion protein knockout neurons and glia. J Neurochem 1998; 70(4): 1686– 1693.
53. Brown DR, Schulz‑ Schaeffer WJ, Schmidt B, Kretzschmar HA. Prion protein‑deficient cells show altered response to oxidative stress due to decreased SOD‑ 1 activity. Exp Neurol 1997; 146(1): 104– 112.
54. Maglio LE, Perez MF, Martins VR, Brentani RR, Ramirez OA. Hippocampal synaptic plasticity in mice devoid of cellular prion protein. Brain Res Mol Brain Res 2004; 131(1– 2): 58– 64.
55. Coitinho AS, Freitas ARO, Lopes MH, Hajj GN, Roesler R, Walz R et al. The interaction between prion protein and laminin modulates memory consolidation. Eur J Neurosci 2006; 24(11): 3255– 3264.
56. Hobzova K, Janouskova O. Tkáňové kultury pro studium prionových chorob. Cesk Slov Neurol N 2010; 73/ 106(4): 379– 386.
57. Steele AD, Lindquist S, Aguzzi A. The prion protein knockout mouse: a phenotype under challenge. Prion 2007; 1(2): 83– 93.
58. Bueler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M et al. Mice devoid of PrP are resistant to scrapie. Cell 1993; 73(7): 1339– 1347.
59. Bueler H, Fischer M, Lang Y, Bluethmann H, Lipp HP, Dearmond SJ et al. Normal development and behavior of mice lacking the neuronal cell‑ surface PrP protein. Nature 1992; 356(6370): 577– 582.
60. Tremblay P, Bouzamondo‑ Bernstein E, Heinrich C, Prusiner SB, Dearmond SJ. Developmental expression of PrP in the post‑implantation embryo. Brain Res 2007; 1139: 60– 67.
61. Hajj GN, Santos TG, Cook ZS, Martins VR. Developmental expression of prion protein and its ligands stress‑ inducible protein 1 and vitronectin. J Comp Neurol 2009; 517(3): 371– 384. doi: 10.1002/ cne.22157.
62. Peralta OA, Huckle WR, Eyestone WH. Developmental expression of the cellular prion protein (PrP(C)) in bovine embryos. Mol Reprod Dev 2012; 79(7): 488– 498. doi: 10.1002/ mrd.22057.
63. Peralta OA, Huckle WR, Eyestone WH. Expression and knockdown of cellular prion protein (PrPC) in differentiating mouse embryonic stem cells. Differentiation 2011; 81(1): 68– 77. doi: 10.1016/ j.diff.2010.09.181.
64. Miranda A, Pericuesta E, Angel Ramirez M, Gutierrez‑ Adan A. Prion protein expression regulates embryonic stem cell pluripotency and differentiation. PLoS ONE 2011; 6(4): e18422. doi: 10.1371/ journal.pone.0018422.
65. Lee YJ, Baskakov IV. Treatment with normal prion protein delays differentiation and helps to maintain high proliferation activity in human embryonic stem cells. J Neurochem 2010; 114(2): 362– 373. doi: 10.1111/ j.1471‑ 4159.2010.06601.x.
66. Lee YJ, Baskakov IV. The cellular form of the prion protein is involved in controlling cell cycle dynamics, self‑ renewal, and the fate of human embryonic stem cell differentiation. J Neurochem 2013; 124(3): 310– 322. doi: 10.1111/ j.1471‑ 4159.2012.07913.x.
67. Adle‑ Biassette H, Verney C, Peoc‘h K, Dauge MC, Razavi F,Choudat L et al. Immunohistochemical expression of prion protein (PrPC) in the human forebrain during development. J Neuropathol Exp Neurol 2006; 65(7): 698– 706.
68. Steele AD, Emsley JG, Ozdinler PH, Lindquist S, Macklis JD. Prion protein (PrPc) positively regulates neural precursor proliferation during developmental and adult mammalian neurogenesis. Proc Natl Acad Sci U S A 2006; 103(9): 3416– 3421.
69. Gage FH. Mammalian neural stem cells. Science 2000; 287(5457): 1433– 1438.
70. Witusik M, Gresner SM, Hulas‑ Bigoszewska K, Krynska B,Azizi SA, Liberski PP et al. Neuronal and astrocytic cells, obtained after differentiation of human neural GFAP‑ positive progenitors, present heterogeneous expression of PrPc. Brain Res 2007; 1186: 65– 73.
71. Toni N, Laplagne DA, Zhao C, Lombardi G, Ribak CE, Gage FH et al. Neurons born in the adult dentate gyrus form functional synapses with target cells. Nat Neurosci 2008; 11(8): 901– 907. doi: 10.1038/ nn.2156.
72. Peretto P, Giachino C, Aimar P, Fasolo A, Bonfanti L. Chain formation and glial tube assembly in the shift from neonatal to adult subventricular zone of the rodent forebrain. J Comp Neurol 2005; 487(4): 407– 427.
73. Relano‑ Gines A, Gabelle A, Hamela C, Belondrade M,Casanova D, Mourton‑ Gilles C et al. Prion replication occurs in endogenous adult neural stem cells and alters their neuronal fate: Involvement of endogenous neural stem cells in prion diseases. PLoS Pathog 2013; 9(8): e1003485.
74. Peng HM, Chen G. Neural precursors derived from human embryonic stem cells. Sci China C Life Sci 2005; 48(3): 295– 299.
75. Kanaani J, Prusiner SB, Diacovo J, Baekkeskov S, Legname G. Recombinant prion protein induces rapid polarization and development of synapses in embryonic rat hippocampal neurons in vitro. J Neurochem 2005; 95(5): 1373– 1386.
76. Bribian A, Fontana X, Llorens F, Gavin R, Reina M, Manuel Garcia‑ Verdugo J et al. Role of the cellular prion protein in oligodendrocyte precursor cell proliferation and differentiation in the developing and adult mouse CNS. PLoS ONE 2012; 7(4): e33872. doi: 10.1371/ journal.pone.0033872.
77. Hartmann CA, Martins VR, Souza Lima FR. High levels of cellular prion protein improve astrocyte development. FEBS Lett 2013; 587(2): 238– 244. doi: 10.1016/ j.febslet.2012.11.032.
78. Kovacs GG, Preusser M, Strohschneider M, Budka H.Subcellular localization of disease‑associated prion protein in the human brain. Am J Pathol 2005; 166(1): 287– 294.
79. Ferrer I. Synaptic pathology and cell death in the cerebellum in Creuzfeldt‑ Jakob disease. Cerebellum 2002; 1(3): 213– 222.
80. Faustino RS, Behfar A, Perez‑ Terzic C, Terzic A. Genomic chart guiding embryonic stem cell cardiopoiesis. Genome Biol 2008; 9(1): R6. doi: 10.1186/ gb‑ 2008‑ 9‑ 1‑ r6.
81. Hidaka K, Shirai M, Lee J‑ K, Wakayama T, Kodama I, Schneider MD et al. The cellular prion protein identifies bipotential cardiomyogenic progenitors. Circ Res 2010; 106(1): 111– 119.
82. Massimino ML, Ferrari J, Sorgato MC, Bertoli A. Heterogeneous PrPC metabolism in skeletal muscle cells. FEBS Lett 2006; 580(3): 878– 884.
83. Stella R, Massimino ML, Sandri M, Sorgato MC, Bertoli A.Cellular prion protein promotes regeneration of adult muscle tissue. Mol Cell Biol 2010; 30(20): 4864– 4876. doi: 10.1128/ MCB.01040‑ 09.
84. Holada K, Vostal JG. Different levels of prion protein (PrPc) expression on hamster, mouse and human blood cells. Br J Haematol 2000; 110(2): 472– 480.
85. Holada K, Simak J, Brown P, Vostal JG. Divergent expression of cellular prion protein on blood cells of human and nonhuman primates. Transfusion 2007; 47(12): 2223– 2232.
86. Holada K, Simak J, Vostal JG. Transmission of BSE by blood transfusion. Lancet 2000; 356(9243): 1772.
87. Lin T, Li RL, Wong BS, Liu DC, Pan T, Petersen RB et al. Normal cellular prior protein is preferentially expressed on subpopulations of murine hemopoietic cells. J Immunol 2001; 166(6): 3733– 3742.
88. Dodelet VC, Cashman NR. Prion protein expression in human leukocyte differentiation. Blood 1998; 91(5): 1556– 1561.
89. Panigaj M, Glier H, Wildova M, Holada K. Expression of prion protein in mouse erythroid progenitors and differentiating murine erythroleukemia cells. PLoS ONE 2011; 6(9): e24599. doi: 10.1371/ journal.pone.0024599.
90. Panigaj M, Brouckova A, Glierova H, Dvorakova E, Simak J, Vostal JG et al. Underestimation of the expression of cellular prion protein on human red blood cells. Transfusion 2011; 51(5): 1012– 1021. doi: 10.1111/ j.1537‑ 2995.2010.02924.x.
91. Zhang CC, Steele AD, Lindquist S, Lodish HF. Prion protein is expressed on long‑term repopulating hematopoietic stem cells and is important for their self‑ renewal. Proc Natl Acad Sci U S A 2006; 103(7): 2184– 2189.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2015 Číslo 4
Nejčtenější v tomto čísle
- Léčba pudendální neuralgie – klinické zkušenosti po pěti letech
- Význam magnetické rezonance v diagnostice epilepsie
- Experimentální léčba poranění míchy
- Léčba foraminálního výhřezu meziobratlové ploténky u istmické spondylolistézy technikou TLIF